




Abstract
The pro-inflammatory cytokine interleukin-6 (IL-6) together with its soluble receptor (sIL-6R) induces and maintains thermal hyperalgesia. It facilitates the heat-induced release of calcitonin gene-related peptide from rat cutaneous nociceptors in vivo and in vitro. Here we report that exposure of nociceptive neurons to the IL-6–sIL-6R complex or the gp130-stimulating designer IL-6–sIL-6R fusion protein Hyper-IL-6 (HIL-6) resulted in a potentiation of heat-activated inward currents (Iheat) and a shift of activation thresholds towards lower temperatures without affecting intracellular calcium levels. The Janus tyrosine kinase inhibitor AG490, the selective protein kinase C (PKC) inhibitor, bisindolylmaleimide 1 (BIM1), as well as rottlerin, a selective blocker of the PKCδ isoform, but not the cyclooxygenase inhibitor indomethacin, effectively reduced the effect. Reverse transcription–polymerase chain reaction (RT–PCR) and in situ hybridization revealed expression of mRNA for the signal-transducing β subunit of the receptor gp130 in neuronal somata, rather than satellite cells in rat dorsal root ganglia. Together, the results suggest that IL-6–sIL-6R acts directly on sensory neurons. It increases their susceptibility to noxious heat via the gp130/Jak/PKCδ signalling pathway.
Introduction
Production of the pleiotropic cytokine interleukin-6 (IL-6) is upregulated, and increased IL-6 serum levels are detected in patients suffering from painful neuropathy (Proietti et al., 1999) malignant tumour or chronic inflammation, all characterized by the most intractable pain and hyperalgesia (Kiefer et al., 2001; Smith et al., 2001).
Most experimental studies reported a pro-inflammatory and pro-nociceptive role for IL-6. Intraplantar, intracerebroventricular or intrathecal injection of IL-6 induced hyperalgesia and/or allodynia in rats (Oka et al., 1995; Poole et al., 1995; DeLeo et al., 1996). In neuropathic mice, nerve injury correlated well with upregulated IL-6 levels and development of thermal hyperalgesia and allodynia (Murphy et al., 1999; DeLeo et al., 1996; Okamoto et al., 2001). IL-6 −/− mice, after carrageenan inflammation or nerve constriction, presented with reduced thermal hyperalgesia (Xu et al., 1997; Murphy et al., 1999). Antisera neutralizing endogenous IL-6 inhibited lipopolysaccharide (LPS)-induced hyperalgesia (Ferreira et al., 1993).
The neuronal effects of IL-6 depended on the presence of soluble IL-6 receptor (sIL-6R) (Marz et al., 1999). All cells in the body express the signalling receptor subunit gp130 but only some cells express a cognate IL-6R which is needed for IL-6 receptor complex formation and signal initiation. The naturally occurring sIL-6R in complex with IL-6 can bind to and activate cells which only express gp130. Therefore, trans-signalling expands the spectrum of cells which are responsive to IL-6. For many neural cells, it has been shown that IL-6 responses depend on trans-signalling. The IL-6–sIL-6R complex acts agonistically on cells that express the signal transducer molecule gp130 as the signal-transducing β subunit of the IL-6 receptor heterotrimer (Taga et al., 1989; Rose-John and Heinrich, 1994). Previous in vitro and in vivo studies demonstrate that short exposure to the IL-6–sIL-6R complex enhanced calcitonin gene-related peptide (CGRP) release, suggesting a direct action on nociceptive neurons (Opree and Kress, 2000; Obreja et al., 2002). Gardiner and colleagues detected immunoreactivity for the IL-6 signal transducer molecule gp130 in neurons (Gardiner et al., 2002). Hyper-IL-6 (HIL-6), a fusion protein of IL-6 and sIL-6R designed as an efficient experimental tool mimicking the effects of the IL-6–sIL-6R complex (Fischer et al., 1997), facilitated sensory and sympathetic neuron survival and nerve regeneration (Marz et al., 1998; Schafer et al., 1999).
In the present study, we investigated the effects of IL-6 together with sIL-6R, as well as of HIL-6 on isolated sensory neurons from adult rat dorsal root ganglia (DRGs) in culture. Reverse transcription–polymerase chain reaction (RT–PCR) and in situ hybridization, whole-cell voltage-clamp recordings and pharmacological tools are used to address the contribution of gp130 and various protein kinases in the signalling cascade.
Material and methods
Cell culture
Rat neuronal cultures were prepared as previously reported (Kress and Guenther, 1999). Experiments were performed in accordance with the legal requirements of the Regierung von Unterfranken. Briefly, lumbar DRGs were harvested from inbred female Wistar rats (140–180 g) and transferred to Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Germany) supplemented with gentamycin (50 µg/ml, Sigma). After removal of the connective tissue, ganglia were incubated for 75 min in collagenase (0.28 U/ml; Roche Biochemicals, Mannheim, Germany) and for 12 min in trypsin (25 000 U/ml, Sigma). Cells were triturated with a fire-polished Pasteur pipette and finally plated on glass-bottomed Petri dishes coated with poly-l-lysine (200 µg/ml, Sigma). The cultures were maintained in serum-free TNB 100™ medium (Biochrom, Berlin, Germany), supplemented with penicillin and streptomycin (200 U/ml each), l-glutamine (2 mM; all from Life Technologies, Germany) and nerve growth factor (mouse NGF 2.5S, 100 ng/ml; Alomone Labs, Tel Aviv, Israel) in a humid 5% CO2 atmosphere at 37°C for 24–36 h.
Electrophysiology
Using the voltage-clamp configuration of the patch-clamp technique, whole-cell ionic currents were recorded from isolated neurons in external solution (ECS) containing (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose (all from Sigma) and 10 HEPES (Merck, Darmstadt, Germany), at pH 7.3 adjusted with NaOH. Borosilicate electrodes (Science Products, Hofheim, Germany) pulled on a horizontal puller (Sutter Instruments Company, Novato, CA) were filled with internal solution (ICS, in mM): 148 KCl, 4 MgCl2, 2 Na-ATP (all from Sigma), 0.2 Li-GTP (Calbiochem, Bad Soden, Germany), 0.1 Fura-2 (Molecular Probes, Leiden, The Netherlands) and 10 HEPES, at pH 7.3 adjusted with KOH. After filling, electrode resistance was 2–4 MΩ. Neurons were clamped at −80 mV holding potential. Currents were filtered at 1 kHz, sampled at 3 kHz and measured using an Axopatch 200A amplifier, a Digidata 1200 interface and the pClamp6.0 software (Axon Instruments, Foster City, CA). Capsaicin sensitivity was tested by application of 1 µM capsaicin for 3 s at the end of the experimental protocol in all cells. Only capsaicin-sensitive cells were included and all of these were also sensitive to noxious heat. Experiments were performed at room temperature and only one neuron was tested per dish. A seven-barrel system with a common outlet was used for fast drug administration and heat stimulation (Dittert et al., 1998). The application system positioned close to the recorded cell (∼100 µm distance) was used to stimulate isolated cells with identical heat stimuli and with different conditioning test solutions. The opening and closing of the solenoid valves were controlled manually from a switchboard. The voltage command for heating the solutions came from the pulse generator of the Axopatch software. In the whole-cell configuration of the voltage-clamp technique, heat-activated inward currents (Iheat) were elicited by applying ramp-shaped heat stimuli at 60 s intervals (linear temperature increase from 25 to 50°C within 5 s). IL-6 (20 ng/ml), alone or together with sIL-6R (25 ng/ml) and HIL-6 (10 ng/ml), respectively, were used as intermittent conditioning stimuli (100–200 s). In some experiments, HIL-6 was applied in the presence of kinase inhibitors or their negative controls.
Ratiometric Ca2+ measurements
For intracellular calcium measurements, neurons were loaded via the patch pipette with the fluorescent calcium indicator dye Fura-2 (100 µM, see above). Background-corrected fluorescent images were taken with a slow scan CCD camera system with fast monochromator (PTI, NJ) coupled to an Axiovert microscope with 40× fluar oil-immersion objective (Zeiss, Jena, Germany). Fura-2 was excited at 340 and 380 nm wavelengths (λ) with equal exposure time (200 ms) and fluorescence was collected at λ = 420 nm. Intracellular [Ca2+]i was calculated as previously reported (Zeilhofer et al., 1996). Calibration constants obtained in vitro were Rmin = 0.44, Rmax = 8.0 and Keff = 1.2 mM.
RT–PCR
Total mRNA was isolated from adult female rat DRGs using RNAzol reagent (WAK-Chemie, Bad-Soden, Germany), then reverse transcribed into cDNA using MuLV reverse transcriptase (Perkin-Elmer Biosystems, Weiterstadt, Germany) as previously described (Haberberger et al., 2000). PCR was performed in a 50 µl reaction volume containing: 1× PCR buffer, 1.5 mM MgCl2, 150 µM dNTP, 0.3 µM of each primer and 1.25 U of AmpliTaq Gold (Applied Biosystems). The amplification conditions were set as follows: initial denaturation at 94°C for 5 min (one cycle); 94°C for 45 s, 58°C for 30 s and 72°C for 45 s (35 cycles), followed by extension at 72°C for 7 min. The gene-specific forward and reverse primers for gp130 as well as Janus kinases (all from Hybaid Interactive Biotechnologie, Ulm, Germany) are specified in Table 1.
Gene-specific primer sequences
| Forward primer | Reverse primer | |
|---|---|---|
| Gp130 | 5′-GGCTCTGAGTCCTTGAAGGCGTACC-3′ | 5′-AACAAGACGCCCAGCAGGGTTG-3′ |
| Jak1 | 5′-CCTGCATGGCTCTGTTGACC-3′ | 5′-CTCATCATGCTGGCTGCCTC-3′ |
| Jak2 | 5′-AGTGCGTGCGAGCGAAGATC-3′ | 5′-AGACCTTCCCGTCCTGCTTG-3′ |
| Jak3 | 5′-CACACCTGGCATCCCGAATC-3′ | 5′-AGCAGTAGGCGGTCGTTGTG-3′ |
| mTyk2 | 5′-CCTGGCCATGACCTGAACAG-3′ | 5′-TGTGCCCTTCACTGACGGAG-3′ |
| Forward primer | Reverse primer | |
|---|---|---|
| Gp130 | 5′-GGCTCTGAGTCCTTGAAGGCGTACC-3′ | 5′-AACAAGACGCCCAGCAGGGTTG-3′ |
| Jak1 | 5′-CCTGCATGGCTCTGTTGACC-3′ | 5′-CTCATCATGCTGGCTGCCTC-3′ |
| Jak2 | 5′-AGTGCGTGCGAGCGAAGATC-3′ | 5′-AGACCTTCCCGTCCTGCTTG-3′ |
| Jak3 | 5′-CACACCTGGCATCCCGAATC-3′ | 5′-AGCAGTAGGCGGTCGTTGTG-3′ |
| mTyk2 | 5′-CCTGGCCATGACCTGAACAG-3′ | 5′-TGTGCCCTTCACTGACGGAG-3′ |
Gene-specific primer sequences
| Forward primer | Reverse primer | |
|---|---|---|
| Gp130 | 5′-GGCTCTGAGTCCTTGAAGGCGTACC-3′ | 5′-AACAAGACGCCCAGCAGGGTTG-3′ |
| Jak1 | 5′-CCTGCATGGCTCTGTTGACC-3′ | 5′-CTCATCATGCTGGCTGCCTC-3′ |
| Jak2 | 5′-AGTGCGTGCGAGCGAAGATC-3′ | 5′-AGACCTTCCCGTCCTGCTTG-3′ |
| Jak3 | 5′-CACACCTGGCATCCCGAATC-3′ | 5′-AGCAGTAGGCGGTCGTTGTG-3′ |
| mTyk2 | 5′-CCTGGCCATGACCTGAACAG-3′ | 5′-TGTGCCCTTCACTGACGGAG-3′ |
| Forward primer | Reverse primer | |
|---|---|---|
| Gp130 | 5′-GGCTCTGAGTCCTTGAAGGCGTACC-3′ | 5′-AACAAGACGCCCAGCAGGGTTG-3′ |
| Jak1 | 5′-CCTGCATGGCTCTGTTGACC-3′ | 5′-CTCATCATGCTGGCTGCCTC-3′ |
| Jak2 | 5′-AGTGCGTGCGAGCGAAGATC-3′ | 5′-AGACCTTCCCGTCCTGCTTG-3′ |
| Jak3 | 5′-CACACCTGGCATCCCGAATC-3′ | 5′-AGCAGTAGGCGGTCGTTGTG-3′ |
| mTyk2 | 5′-CCTGGCCATGACCTGAACAG-3′ | 5′-TGTGCCCTTCACTGACGGAG-3′ |
The amplified fragments were cloned into TOPO vector (Invitrogen GmbH, Karlsruhe, Germany) and sequenced on the Applied Biosystems 373 DNA sequencer using Taq Dye Deoxy Terminator cycle sequencing kits (Perkin-Elmer Biosystems) to confirm the identity of the amplified products.
TaqMan RT–PCR analysis
For analysis of mRNA levels, total RNA was isolated from DRG neurons immediately after dissociation and purification with bovine serum albumin (Sigma) by using the NucleoSpin RNA II kit (Macherey-Nagel). Reverse transcription to cDNA was carried out using the GeneAmp RNA PCR kit (Applied Biosystems) according to the manufacturer's instructions. For each sample, 20 ng of cDNA and five times 1 : 3 serial dilutions thereof were analysed for expression of rat leukaemia inhibitory factor receptor (LIFR), IL-6R, gp130 and actin by real-time quantitative RT–PCR using the fluorescent TaqMan 5′-nuclease assay as previously described (Ludwig et al., 2002). The TaqMan real time-PCR was performed using 2× TaqMan Master Mix (Applied Biosystems) and 20× assay-on-demand TaqMan primers and probes in a total volume of 20 µl. The mRNA level for the gene of interest was quantified as the percentage of that determined for β-actin. For each PCR, the cycle threshold (CT) values for the gene of interest and β-actin were determined. From these, ΔCT values were calculated as CTactin − CTgene of interest and results were then expressed as 2ΔCT × 100. TaqMan primers and probes were obtained from Applied Biosystems and had the following identification numbers: LIFR, Rn00579104_m1; IL-6R, Rn00566707_m1; gp130, Rn01489667_m1; β-actin, Rn00667869_m1. The primers were intron spanning and therefore did not amplify significant amounts of genomic cDNA. The correct size of the amplified cDNA was controlled by agarose gel electrophoresis.
In situ hybridization
The gp130 PCR product (see above), T7 RNA polymerase and DIG labelling mix (Roche Biochemicals, Mannheim, Germany) were used to produce digoxigenin (DIG)-labelled antisense and sense RNA-probes. Lumbar DRGs were shock frozen in isopentane and then cooled in liquid N2. Cryosections (10 µM thick) were cut, fixed (4% phosphate-buffered paraformaldehyde), permeabilized (0.01 M sodium citrate 10 min in a microwave at 250 W; 0.2 M HCl for 20 min; phosphate-buffered 0.3% Triton X-100 for 5 min; 2 µg/ml proteinase K from Sigma for 20 min, 37°C) and acetylated [0.1 M triethanolamine containing 0.252% (v/v) acetic anhydride, 10 min, rapid stirring]. Pre-hybridization was done using: 2.5% 50× Denhardt's, 0.05 M EDTA, 0.5 mg/ml yeast tRNA in 50 mM Tris–HCl for 2 h at 45°C. The slices were then incubated with 10 µg/ml probe in 0.1 M Tris–HCl, 50% deionized formamide, 0.05 M EDTA, 0.25 mg/ml yeast tRNA, 0.5 mg/ml herring sperm DNA, 25% dithiothreitol, 0.002% NaCl and 10% dextran sulphate (12–16 h, 45°C). Sections were washed in standard sodium citrate buffer (2× and 1×, 20 min each); 20 µg/ml RNase A (Sigma, 30 min, 37°C); decreasing concentrations of standard citrate buffer (1× and 0.5×, 20 min each and 0.2×, 20 min at 24°C, 1 h at 50°C and finally 20 min at 24°C); distilled water (5 min); 0.1 M Tris–HCl (10 min) and 0.1 M maleate buffer (10 min). Detection of the DIG-labelled probe was performed as recommended by the manufacturer, with alkaline phosphate-conjugated DIG antibody (4°C, 12 h). Colour development was allowed to proceed in the darkness for 4–16 h. The reaction was terminated by immersion in PBS (pH 7.5). Sections were mounted with Keyser's glycerylgelatine (Merck, Darmstadt, Germany). Adjacent control sections from the same ganglia were incubated with sense probes and otherwise treated equally.
Reagents
IL-6 was purchased from Roche Molecular Biochemicals, Mannheim, Germany and sIL-6R was from R&D Systems, Wiesbaden-Nordenstadt, Germany. HIL-6 was synthesized as previously described (Fischer et al., 1997). Bisindolylmaleimide I (BIM1) and rottlerin were both obtained from Sigma. Bisindolylmaleimide V (BIM5), AG490 and Jak3-specific inhibitor were purchased from Calbiochem, San Diego, CA. Aliquots of stock solutions were prepared in distilled water or DMSO (final DMSO concentration ≤0.1%) and diluted in external solution (ECS) to the final concentration before the experiment.
Statistical evaluation
For ionic current analysis, we used Origin 7.0 (Origin Lab Corporation, Northampton, MA). After leak subtraction, mean peak Iheat amplitudes before and after cytokine stimulation were measured. For data analysis, the Statistica software package StatSoft 5.1 (Tulsa, USA) was used. All summarizing values are given as means ± SEM. Data were normalized by dividing the current value immediately after the conditioning stimulus by the current value just before the stimulus. To analyse the heat threshold, current shifts were plotted against temperature for each neuron and the temperature threshold after stimulation was subtracted from the one before stimulation. The Mann–Whitney U test was used for inter-individual comparisons and the Wilcoxon matched pairs test was calculated for intra-individual comparisons. Differences were considered significant at P < 0.05.
Results
IL-6 together with sIL-6R potentiates heat-activated ionic currents in sensory neurons
Under the conditions used, Iheat neither sensitized nor desensitized with repetitive thermal stimulation at 1 min intervals. IL-6 or the IL-6–sIL-6R complex affected neither the leak conductance nor the access resistance, and no inward or outward current was elicited nor was a change in intracellular calcium levels observed during the application of the cytokine (Fig. 1A). IL-6 and sIL-6R (n = 3) alone were completely ineffective. As shown in Fig. 1, Iheat before and after IL-6 application (20 ng/ml) were very similar (447 ± 67 pA before versus 481 ± 72 pA after IL-6, n = 10). In four other cells, even a higher concentration of IL-6 (200 ng/ml) did not affect the Iheat (data not shown).
![IL-6 together with sIL-6R potentiates heat-activated ionic currents in sensory neurons. (A) Example of a neuron showing no change of heat-activated currents (Iheat) after exposure to IL-6 (20 ng/ml). However, facilitation of Iheat was observed when the same neuron was subsequently exposed to IL-6 together with sIL-6R (25 ng/ml). Iheat were elicited by identical 5 s ramp-shaped heat stimuli (H1–H5) at 1 min intervals. The lower panel depicts the intracellular calcium concentration [Ca2+]i. Neither IL-6 nor IL-6–sIL-6R treatment yielded any [Ca2+]i increase in the tested neuron. (B) Mean ± SEM of Iheat amplitudes before and after exposure to IL-6 (20 ng/ml) (∼100 s; n = 10). (C) Mean ± SEM of Iheat amplitudes before and after exposure to IL-6 (20 ng/ml)–sIL-6R (25 ng/ml) (∼100 s; n = 6). The asterisk marks significant differences (*P < 0.05).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/128/7/10.1093/brain/awh490/2/m_awh490f1.gif?Expires=1716401944&Signature=lLYbjB45EQeBfcrDTRtB61zxG9nw8lehpJuAunHCAE9Y8LoiwtmS70lU4UaYj8NyzoAaPfVMSW65DBbw-WA6RXPPQICNqU0unE4Z1BYGwlZLSXMkTb2w5fALNYb3A1LQ61WdHEfGrhoxwTIu9IcHQujNtbS9BhxOzKHNszvj72zzH0tZiYJdYFuDwwHvyG1ScE31VGDmT29UuWRQMmhwofn94lKncoE1iDBnw5W67QgGZHsI0puno7h2w9s-cUMpkfiN8I5GeHLoORe~caidYZ9t-NQiRxcoEaA7lxJVM4jhiRDdWTEhR59KdevjVjwuvmFDxrkafMs-dfxrouRWhA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IL-6 together with sIL-6R potentiates heat-activated ionic currents in sensory neurons. (A) Example of a neuron showing no change of heat-activated currents (Iheat) after exposure to IL-6 (20 ng/ml). However, facilitation of Iheat was observed when the same neuron was subsequently exposed to IL-6 together with sIL-6R (25 ng/ml). Iheat were elicited by identical 5 s ramp-shaped heat stimuli (H1–H5) at 1 min intervals. The lower panel depicts the intracellular calcium concentration [Ca2+]i. Neither IL-6 nor IL-6–sIL-6R treatment yielded any [Ca2+]i increase in the tested neuron. (B) Mean ± SEM of Iheat amplitudes before and after exposure to IL-6 (20 ng/ml) (∼100 s; n = 10). (C) Mean ± SEM of Iheat amplitudes before and after exposure to IL-6 (20 ng/ml)–sIL-6R (25 ng/ml) (∼100 s; n = 6). The asterisk marks significant differences (*P < 0.05).
When applied together with sIL-6R (25 ng/ml), IL-6 significantly potentiated Iheat amplitudes and, in six out of 11 neurons (54.5%), Iheat increased from 466 ± 71 to 824 ± 143 pA (P < 0.05; Fig. 1C). In four of six other neurons (66.7%), a higher dose of IL-6 (200 ng/ml) together with sIL-6R (25 ng/ml) caused a similar increase of Iheat peak current from 428 ± 60 to 853 ± 141 pA (data not shown).
Expression of gp130 mRNA in rat sensory neurons
RT–PCR revealed gp130 mRNA expression in rat DRGs (Fig. 2A). To determine whether gp130 mRNA was located in neurons or in non-neuronal cells, non-radioactive in situ hybridization with sequence-specific antisense and sense probes was performed in frozen DRG sections. Gp130 mRNA was detected in all neurons, including small and medium size ones, whereas it was not detectable in non-neuronal cells (Fig. 2B and D). No signal was detectable in sections treated with the sense probes as negative controls (Fig. 2C). In contrast to gp130 and the ligand-binding cytokine receptor subunit for LIFR, another member of the gp130-stimulating cytokine family, which were abundantly expressed in DRGs, mRNA levels for the membrane-bound IL-6 receptor were >50-fold lower as compared with gp130 or LIFR mRNA (Fig. 2E; P < 0.05, n = 3)
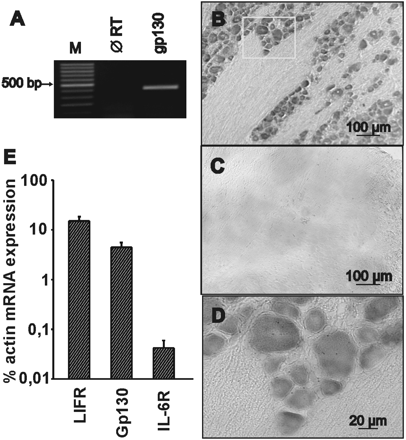
The signal transducer molecule gp130 is expressed in rat DRGs. (A) RT–PCR reveals the expression of gp130 mRNA in rat DRGs (M, marker; ∅ RT, without reverse transcriptase). (B and D) In situ hybridization with gp130-specific antisense probes shows the neuronal expression of gp130 mRNA in rat DRG sections. (C) Negative control with sense probes. (E) Relative expression of IL-6R, LIFR and gp130 mRNA in rat DRG samples. Preparations of mRNA from rat DRG were analysed for the expression level of IL6R, LIFR, gp130 and β-actin mRNA using TaqMan RT–PCR. For each gene, PCRs were carried out with serial dilutions of cDNA. The mean expression levels (±SD) of IL-6R, LIFR and gp130 mRNA in DRG of three different animals were calculated as the percentage of actin mRNA expression.
IL-6-induced sensitization does not depend on prostaglandin production
The fusion protein HIL-6 has been designed as a potent experimental tool to stimulate gp130 and evoke IL-6–sIL-6R effects (Fischer et al., 1997). In our experiments, during HIL-6 application, neither ionic currents nor rises in intracellular calcium concentration were detected (Fig. 3A). Similar to the IL-6–sIL-6R complex, HIL-6 (10 ng/ml) significantly increased Iheat peak currents in 10 out of 15 neurons (66.67%) from 392 ± 50 to 913 ± 81 pA (P < 0.01, Fig. 3). Current versus temperature plots revealed a drop in the heat activation thresholds after HIL-6 treatment accompanying the peak current increase. In the neuron depicted as a representative example in Fig. 3C, the threshold dropped from 46.5 to 42.5°C after exposure to HIL-6. In all responsive neurons, a similar drop of heat thresholds was observed after both IL-6–sIL-6R and HIL-6 treatment as compared with IL-6 alone (2.37 ± 0.74°C after IL-6–sIL-6R; 2.18 ± 0.24°C after HIL-6; 0.23 ± 0.33°C after IL-6, P < 0.01 and P < 0.001, respectively; Fig. 3D). HIL-6 exhibited a bell-shaped dose–response relationship with a maximum effect at concentrations between 1 and 5 ng/ml and an EC50 of 200 pg/ml. This suggests that the shift in Iheat occurs only within a small range of IL-6–sIL-6R complexes (Fig. 3E).
![HIL-6 sensitizes sensory neurons to noxious heat via a prostaglandin-independent pathway. (A) Example of a neuron showing facilitation of heat-activated currents (Iheat) after exposure to HIL-6 (10 ng/ml). Iheat were elicited by identical 5 s ramp-shaped heat stimuli (H1–H5) at 1 min intervals. HIL-6 treatment did not change [Ca2+]i levels (lower panel). (B) Mean ± SEM of Iheat amplitudes before and after exposure to HIL-6 (10 ng/ml) (∼100 s; n = 10). (C) Example of a neuron showing a drop in the heat threshold from 46.5 to 42.5°C after exposure to HIL-6 (10 ng/ml). (D) Mean ± SEM decrease in heat thresholds after IL-6, IL-6–sIL-6R or HIL-6 treatment. The numbers in parentheses represent numbers of neurons tested. Asterisks mark significant differences (*P < 0.05; **P < 0.01; ***P < 0.001). The dose–response relationship for HIL-6 is given in E; the numbers of cells for each concentration range between 4 and 5. (F) Mean ± SEM. Iheat increase after HIL-6 (10 ng/ml) or HIL-6 + indomethacin (30 µM). The numbers in parentheses represent the numbers of neurons tested.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/128/7/10.1093/brain/awh490/2/m_awh490f3.gif?Expires=1716401944&Signature=TivL2w1uIVtCerRuRdy5oYJAII27m2oB0w3Rnei0onrS1p-hI1tYyZQA67mMtg1MrnvZePDJsQyc0oK9bTdTw5dCaOxzv3XXBH5Ks2HU28kZC41D2Ntt7Yk~LXArlS-9UsHY-uBdMaNeU9KESg7DE3vcFOdqnXYKvV02yEfZqI0aiV5Q6MeK7DgYxMIUidumrFFGpieY71WilAaD132Hkuhf8n0HAS8D17Qbuz4Xl53iNc1-ZKtBn7PBkxJqGCF5ncWccF8jVkWVD1YXIdt1bDcmMQfqk1mkXOZl6R8uh0C9jJdRtefS13XzuAAQnODXtNIzIY-J1CN-sECXQ~kEdw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HIL-6 sensitizes sensory neurons to noxious heat via a prostaglandin-independent pathway. (A) Example of a neuron showing facilitation of heat-activated currents (Iheat) after exposure to HIL-6 (10 ng/ml). Iheat were elicited by identical 5 s ramp-shaped heat stimuli (H1–H5) at 1 min intervals. HIL-6 treatment did not change [Ca2+]i levels (lower panel). (B) Mean ± SEM of Iheat amplitudes before and after exposure to HIL-6 (10 ng/ml) (∼100 s; n = 10). (C) Example of a neuron showing a drop in the heat threshold from 46.5 to 42.5°C after exposure to HIL-6 (10 ng/ml). (D) Mean ± SEM decrease in heat thresholds after IL-6, IL-6–sIL-6R or HIL-6 treatment. The numbers in parentheses represent numbers of neurons tested. Asterisks mark significant differences (*P < 0.05; **P < 0.01; ***P < 0.001). The dose–response relationship for HIL-6 is given in E; the numbers of cells for each concentration range between 4 and 5. (F) Mean ± SEM. Iheat increase after HIL-6 (10 ng/ml) or HIL-6 + indomethacin (30 µM). The numbers in parentheses represent the numbers of neurons tested.
Previously, IL-6 has been reported to activate phospholipase A2 rapidly with subsequent prostaglandin formation (Wu et al., 2002). Therefore, a prostanoid-dependent autocrine effect might be responsible for the actual HIL-6-induced heat sensitization. To address this issue, some experiments were performed in the presence of the cyclooxygenase inhibitor indomethacin (30 µM). However, our results clearly show a similar HIL-6-induced potentiation of Iheat in the presence of indomethacin as compared with controls (2.4-fold for HIL-6 with indomethacin versus a 2.5-fold increase for HIL-6 without indomethacin; n = 6, Fig. 3F). This demonstrates that an autocrine prostaglandin action does not contribute to the present HIL-6 effects.
Involvement of Jak tyrosine kinases
In many models, Janus kinases were involved at very early stages of IL-6 signalling. RT–PCR revealed mRNA expression for all members of the Janus kinase family except for Tyk2 in primary neuronal cultures from rat DRGs (Fig. 4A). Therefore, we performed electrophysiological experiments in the presence of the Jak inhibitor AG490. Under these conditions, HIL-6-induced sensitization was inhibited in a concentration-dependent manner (1.2-fold with 10 µM AG490 versus a 2.5-fold increase after HIL-6 alone, P < 0.01, IC50 = 90.16 nM, Fig. 4). However, AG490 non-selectively inhibits Jaks including Jak3 that does not contribute to IL-6 signalling. We therefore used as control a specific Jak3 inhibitor (10 µM) which did not prevent HIL-6-induced Iheat potentiation (from 538.2 ± 65 to 1053.4 117.1 pA; n = 5; P < 0.05).
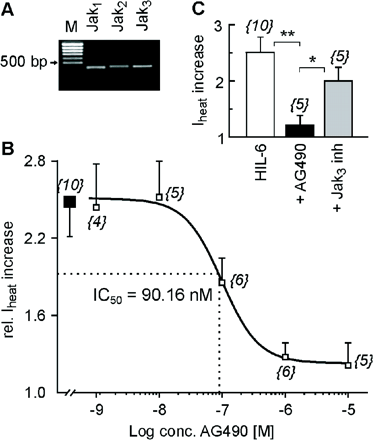
IL-6-induced sensitization of Iheat depends on Jak activation. (A) RT–PCR reveals the expression of the Janus kinases Jak1–3 mRNA in lumbar rat DRGs (M = marker). (B) Concentration–response curve for the inhibitory effect of the Jak inhibitor AG490 (open squares) on the relative HIL-6-induced Iheat increase (filled square): IC50 = 90.16 nM. The numbers in parentheses correspond to the numbers of neurons tested. (C) Mean ± SEM Iheat increase after HIL-6 (10 ng/ml), HIL-6 + AG490 (10 µM) or HIL-6 + Jak3 inhibitor (10 µM). Responses were normalized to the current amplitude immediately preceding HIL-6 application. The numbers in parentheses correspond to the number of neurons tested. Asterisks mark significant differences (*P < 0.05; **P < 0.01).
Involvement of protein kinase Cδ
Downstream of Jak, protein kinase C (PKC) can be recruited when gp130 signal transduction is initiated. In our experiments, the selective PKC inhibitor BIM1 (200 nM) prevented the HIL-6-induced potentiation of Iheat peak currents (346 ± 36 pA before versus 398 ± 15 pA after HIL-6) and the accompanying drop in heat activation threshold (0.26 ± 0.41°C) (n = 5, Fig. 5). In contrast, the negative control BIM5 had no effect. Recent data suggest a specific IL-6-dependent interaction between gp130 and PKCδ. We therefore performed current recordings in the presence of the specific PKCδ inhibitor rottlerin. This compound dose-dependently inhibited the HIL-6-induced potentiation of Iheat in rat sensory neurons (IC50 = 163 nM, Fig. 5).
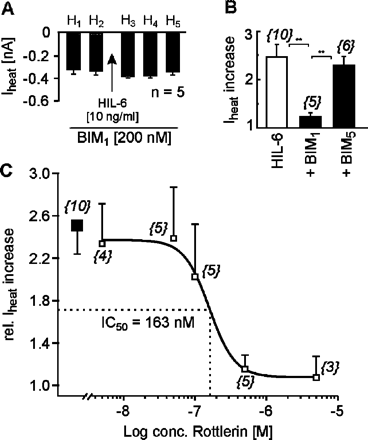
IL-6-induced sensitization of Iheat depends on PKC activation. (A) Mean ± SEM Iheat amplitudes of current responses to heat stimuli (H1–H5) before and after exposure to HIL-6 in the presence of BIM1 (200 nM; n = 5). (B) Mean ± SEM Iheat increase after HIL-6 (10 ng/ml), HIL-6 + BIM1 (200 nM) or HIL-6 + BIM5 (200 nM). Responses were normalized to the current amplitude immediately preceding HIL-6 application. The numbers in parentheses correspond to the numbers of neurons tested. Asterisks mark significant differences (**P < 0.01). (C) Concentration–response curve for the inhibitory effect of the PKCδ inhibitor rottlerin (open squares) on the HIL-6-induced Iheat increase (filled square): IC50 = 163 nM. The numbers in parentheses correspond to the numbers of neurons tested.
Discussion
In the present study, IL-6 when applied together with its soluble receptor potentiated heat-activated ionic currents in capsaicin-sensitive neurons from rat DRGs. The potentiation was inhibited by the Jak inhibitor AG490 but not by selective Jak3 inhibition. Furthermore, the specific PKC inhibitor BIM1, as well as rottlerin which is selective for the PKCδ isoform, prevented IL-6-induced potentiation. RT–PCR revealed expression of mRNA for the signal-transducing β subunit of the IL-6 receptor (gp130) in DRG neurons. Together, the data suggest pro-algesic effects of IL-6 on primary nociceptive afferents and an involvement of Jak tyrosine kinases 1 and 2 as well as PKCδ in IL-6 signalling.
The cytokine IL-6 did not evoke excitatory inward currents, and this corroborates the lack of nociceptor excitation in previous in vitro as well as in vivo studies (Opree and Kress, 2000; Obreja et al., 2002). However, other in vitro studies reported excitation of a small subpopulation (7%) of myenteric neurons, which probably was due to indirect effects (Kelles et al., 2000). In many tissues, IL-6 exerted its effects by binding to a ligand-binding 80 kDa low-affinity component (IL-6R) and a signal-transducing non-ligand binding 130 kDa glycoprotein (gp130) which is also part of the receptor complex for several IL-6-related cytokines and which is expressed at the transcriptional and translational level in sensory neurons of DRGs (Gardiner et al., 2002). Very low levels of sIL-6R, which is released from leukocytes and other cell types by alternative splicing and shedding, were found under physiological conditions in human plasma, urine, CSF and synovial fluid (Novick et al., 1989; Lust et al., 1992; Mullberg et al., 1993; Muller-Newen et al., 1996; Wierzbowska et al., 1999). However, levels of sIL-6R were elevated during, for example, autoimmune diseases, human immunodeficiency virus (HIV) infection or chronic arthritis (Honda et al., 1992; Gaillard et al., 1993; Keul et al., 1998). Cells expressing gp130 but not IL-6R could be stimulated by a complex of IL-6 bound to sIL-6R (Taga et al., 1989; Rose-John and Heinrich, 1994). Neurons expressing gp130 mRNA required sIL-6R in order to respond to IL-6 stimulation, and this is in line with the present study on nociceptive neurons (Marz et al., 1999).
In our experiments, IL-6 induced significant potentiation of Iheat peak amplitudes and a shift of activation thresholds towards lower temperatures which was independent of rises in intracellular calcium concentration. The sensitization process occurred at short latency, and this suggested the involvement of a fast signalling cascade, rather than the classical cytokine-induced changes in gene expression. One possibility would be the receptor-mediated activation of protein kinases and the consecutive phosphorylation of a heat-transducing ion channel, e.g. of the TRPV family. In many cellular models, the binding of the IL-6–sIL-6R complex to gp130 induced activation of receptor-associated Janus kinases (Taga et al., 1989; Murakami et al., 1993). RT–PCR revealed mRNA expression for Jak1, Jak2 and Jak3, but not Tyk2, in rat DRGs. Sensitization of Iheat was inhibited by AG490 rather than by the Jak3 inhibitor. We therefore conclude that Jak1 and Jak2 kinases may be involved at an early stage in the signalling cascade.
Furthermore, PKC was reported to be activated by IL-6/gp130, and recent evidence supports the specific involvement of the calcium-independent ‘novel’ isoform δ (Jain et al., 1999; Novotny-Diermayr et al., 2002). In the present study, IL-6-induced potentiation of Iheat was blocked by the PKC inhibitor BIM1, as well as the selective PKCδ inhibitor rottlerin. This is in line with previous evidence that PKC activation potentiated heat sensitivity of nociceptive neurons (Kumazawa et al., 1991; Cesare and McNaughton, 1996; Leng et al., 1996). A specific role for the ε isoform of PKC was reported for bradykinin-induced Iheat potentiation in sensory neurons (Cesare et al., 1999; Khasar et al., 1999). However, other PKC isoforms detected in DRG neurons including PKCδ similarly increase nociceptor heat sensitivity (Cesare et al., 1999).
Members of the heat-transducing vanilloid receptor family TRPV are regulated by PKC-mediated phosphorylation (Benham et al., 2002). In the present study, all neurons exhibited heat thresholds in a range between 42 and 47°C, similar to thresholds reported for the vanilloid receptor-1 (TRPV1) (Caterina et al., 1997). Recent studies reported regulatory effects of PKC on TRPV1 activity (Premkumar and Ahern, 2000; Vellani et al., 2001; Sugiura et al., 2002), and two PKC phosphorylation sites in the molecular structure of TRPV1 were identified (Numazaki et al., 2002). Therefore, TRPV1 seems to be a likely candidate that may be regulated by IL-6 signalling via PKC-dependent phosphorylation; however, the contribution of other heat-sensitive ion channels, e.g. TRPV3 or TRPV4, cannot be excluded.
Together, our findings suggest that the IL-6–sIL-6R complex but not IL-6 or sIL-6R alone activates gp130 in order to sensitize nociceptors to heat. Janus kinases and PKCδ are involved in downstream signalling which probably leads to the phosphorylation and sensitization of a heat-activated ion channel, possibly TRPV1. This novel mechanism for the first time explains the fast pro-algesic effect of IL-6 in inflamed tissue. In addition, it may help to understand the mechanisms that cause neuropathic pain and hyperalgesia: heat transducer molecules are not only found in the nociceptive nerve terminal but they are also functionally expressed along the axon (Sauer et al., 1999, 2001). Upon nerve injury, immune cells invade the subglial space and may locally release large amounts of cytokines onto the axon and/or cell body (Hu and McLachlan, 2002). This may cause sensitization of nociceptive neurons at sites that normally do not contribute to the detection of thermal stimuli. We suggest that a drop of the heat threshold below body core temperature at these sites would explain ectopic discharge originating from the axon or soma (Devor et al., 1992) and this may essentially contribute to the generation of neuropathic pain and hyperalgesia following nerve injury. Since sensitized heat nociception by IL-6 depended on IL-6–sIL-6R trans-signalling, a novel therapeutic strategy might make use of the soluble gp130 protein from which we have shown that it exclusively inhibits IL-6 trans-signalling without affecting IL-6 responses via the membrane-bound IL-6 responses (Atreya et al., 2000; Jostock et al., 2001).
We wish to thank D. Thierschmidt for excellent technical assistance, and H. O. Handwerker for continuous support. The work was funded by the Deutsche Forschungsgemeinschaft (SFB-353/A10), the Marohn-Stiftung and the Wilhelm Sander-Stiftung (1996.058.2).
References
Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo.
Benham CD, Davis JB, Randall AD. Vanilloid and TRP channels: a family of lipid-gated cation channels.
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway.
Cesare P, McNaughton PA. Novel heat-activated current in nociceptive neurons and its sensitization by bradykinin.
Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat.
DeLeo JA, Colburn RW, Nichols M, Malhotra A. Interleukin-6-mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model.
Devor M, Wall PD, Catalan N. Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction.
Dittert I, Vlachova V, Knotkova H, Vitaskova Z, Vyklicky L, Kress M, et al. A technique for fast application of heated solutions of different composition to cultured neurones.
Ferreira SH, Lorenzetti BB, Poole S Bradykinin initiates cytokine-mediated inflammatory hyperalgesia.
Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A, et al. I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion.
Gaillard JP, Bataille R, Brailly H, Zuber C, Yasukawa K, Attal M, et al. Increased and highly stable levels of functional soluble interleukin-6 receptor in sera of patients with monoclonal gammopathy.
Gardiner NJ, Cafferty WB, Slack SE, Thompson SW. Expression of gp130 and leukaemia inhibitory factor receptor subunits in adult rat sensory neurones: regulation by nerve injury.
Haberberger R, Scholz A, Kummer W, Kress M. M2-receptor subtype does not mediate muscarine-induced increases in [Ca2+](i) in nociceptive neurons of rat dorsal root ganglia.
Honda M, Yamamoto S, Cheng M, Yasukawa K, Suzuki H, Saito T, et al. Human soluble IL-6 receptor: its detection and enhanced release by HIV infection.
Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat.
Jain N, Zhang T, Kee WH, Li W, Cao X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner.
Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transignaling responses.
Kelles A, Janssens J, Tack J. IL-1 beta and IL-6 excite neurones and suppress cholinergic neurotransmission in the myenteric plexus of the guinea pig.
Keul R, Heinrich PC, Muller-Newen G, Muller K, Woo P. A possible role for soluble IL-6 receptor in the pathogenesis of systemic onset juvenile chronic arthritis.
Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice.
Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune-mediated damage to the peripheral nervous system.
Kress M, Guenther S. Role of [Ca2+]i in the ATP-induced heat sensitization process of rat nociceptive neurons.
Kumazawa T, Mizumura K, Minagawa M, Tsujii Y Sensitizing effects of bradykinin on the heat responses of the visceral nociceptor.
Leng S, Mizumura K, Koda H, Kumazawa T. Excitation and sensitization of the heat response induced by a phorbol ester in canine visceral polymodal receptors studied in vitro.
Ludwig A, Berkhout T, Moores K, Groot P, Chapman G. Fractalkine is expressed by smooth muscle cells in response to IFN-gamma and TNF-alpha and is modulated by metalloproteinase activity.
Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor.
Marz P, Cheng JG, Gadient RA, Patterson PH, Stoyan T, Otten U, et al. Sympathetic neurons can produce and respond to interleukin 6.
Marz P, Otten U, Rose-John S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors.
Mullberg J, Schooltink H, Stoyan T, Gunther M, Graeve L, Buse G, et al. The soluble interleukin-6 receptor is generated by shedding.
Muller-Newen G, Kohne C, Keul R, Hemmann U, Muller-Esterl W, Wijdenes J, et al. Purification and characterization of the soluble interleukin-6 receptor from human plasma and identification of an isoform generated through alternative splicing.
Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, et al. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase.
Murphy PG, Ramer MS, Borthwick L, Gauldie J, Richardson PM, Bisby MA. Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice.
Novick D, Engelmann H, Wallach D, Rubinstein M. Soluble cytokine receptors are present in normal human urine.
Novotny-Diermayr V, Zhang T, Gu L, Cao X. Protein kinase C delta associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3–gp130 interaction.
Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues.
Obreja O, Schmelz M, Poole S, Kress M. Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo.
Oka T, Oka K, Hosoi M, Hori T. Intracerebroventricular injection of interleukin-6 induces thermal hyperalgesia in rats.
Okamoto K, Martin DP, Schmelzer JD, Mitsui Y, Low PA. Pro- and anti-inflammatory cytokine gene expression in rat sciatic nerve chronic constriction injury model of neuropathic pain.
Opree A, Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin.
Poole S, Cunha FQ, Selkirk S, Lorenzetti BB, Ferreira SH. Cytokine-mediated inflammatory hyperalgesia limited by interleukin-10.
Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C.
Proietti G, Puliti M, Tulli F, Silvestri M. A man with worsening weakness.
Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function.
Sauer SK, Bove GM, Averbeck B, Reeh PW. Rat peripheral nerve components release calcitonin gene-related peptide and prostaglandin E2 in response to noxious stimuli: evidence that nervi nervorum are nociceptors.
Sauer SK, Reeh PW, Bove GM. Noxious heat-induced CGRP release from rat sciatic nerve axons in vitro.
Schafer KH, Mestres P, Marz P, Rose-John S. The IL-6/sIL-6R fusion protein hyper-IL-6 promotes neurite outgrowth and neuron survival in cultured enteric neurons.
Smith PC, Hobisch A, Lin DL, Culig Z, Keller ET. Interleukin-6 and prostate cancer progression.
Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1.
Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130.
Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide.
Wierzbowska A, Urbanska-Rys H, Robak T. Circulating IL-6-type cytokines and sIL-6R in patients with multiple myeloma.
Wu T, Han C, Lunz JG, III, Michalopoulos G, Shelhamer JH, Demetris AJ. Involvement of 85-kd cytosolic phospholipase A(2) and cyclooxygenase-2 in the proliferation of human cholangiocarcinoma cells.
Xu XJ, Hao JX, Andell-Jonsson S, Poli V, Bartfai T, Wiesenfeld-Hallin Z. Nociceptive responses in interleukin-6-deficient mice to peripheral inflammation and peripheral nerve section.
Author notes
1Institut für Physiologie und Experimentelle Pathophysiologie, Friedrich-Alexander Universität, Erlangen-Nürnberg, 2Institut für Anatomie und Zellbiologie, Justus-Liebig Universität, Giessen, 3Institut für Biochemie, Christian-Albrecht Universität, Kiel, Germany and 4Department für Physiologie und Medizinische Physik, Division Physiologie, Medizinische Universität, Innsbruck, Austria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}