




Abstract
We investigated three separate families (designated D, F and G) with frontotemporal dementia that have the same molecular mutation in exon 10 of the tau gene (P301L). The families share many clinical characteristics, including behavioural aberrations, defective executive functions, language deficits, relatively preserved constructional abilities and frontotemporal atrophy on imaging studies. However, Family D has an earlier mean age of onset and shorter duration of disease than Families F and G (49.0 and 5.1 years versus 61–64 and 7.3–8.0 years, respectively). Two members of Families D and F had neuropathological studies demonstrating lobar atrophy, but the brain from Family D had prominent and diffuse circular, intraneuronal, neurofibrillary tangles not seen in Family F. The brain from Family F had ballooned neurons typical of Pick's disease type B not found in Family D. A second autopsy from Family D showed neurofibrillary tangles in the brainstem with a distribution similar to that found in progressive supranuclear palsy. These three families demonstrate that a missense mutation in the exon 10 microtubule-binding domain of the tau protein gene can produce severe behavioural abnormalities with frontotemporal lobar atrophy and microscopic tau pathology. However, the findings in these families also emphasize that additional unidentified environmental and/or genetic factors must be producing important phenotypic variability on the background of an identical mutation. Apolipoprotein E genotype does not appear to be such a factor influencing age of onset in this disease.
Introduction
Arnold Pick first described behavioural and language abnormalities associated with frontotemporal lobar atrophy in 1892 (Pick, 1892). Nearly 100 years later, Wilhelmsen et al. (1994) showed linkage of disinhibition dementia in a family to genetic markers on chromosome 17. It soon became apparent that several phenotypically different families shared this same linkage relationship and expressed various types of tau pathology, and the disorder was called FTDP-17 (frontotemporal dementia with parkinsonism linked to chromosome 17) (Foster et al., 1997; Spillantini et al., 1998a). Tau became an immediate candidate gene for this disorder because it was located within the critical 17q21–22 region. Poorkaj et al. (1998) Spillantini et al. (1998b) and Hutton et al. (1998) have found several kindreds with a variety of mutations within the tau gene. These findings are of importance to the broad field of neurodegenerative disorders because tau pathology is found in a wide range of conditions including Alzheimer's disease, Pick's disease, progressive supranuclear palsy, Guamanian amyotrophic lateral sclerosis–Parkinson's disease–dementia complex, postencephalitic Parkinson's disease and corticobasal degeneration (Feany and Dickson, 1996). The purpose of the present study was to compare and contrast the clinical and pathological characteristics of three families with frontotemporal dementia having an identical mutation in exon 10 of the tau gene (P301L). These families have important phenotypic differences as well as expected similarities. We were also able to genotype several affected family members for apolipoprotein E alleles, allowing us to evaluate whether the Apo E genotype influenced age of onset as it does for Alzheimer's disease (Roses, 1994).
Method
The three families (D, F and G) were ascertained through the University of Washington and Oregon Health Sciences Alzheimer's Disease Research Centers (ADRCs). Family F was also independently ascertained through the Massachusetts General Hospital ADRC. These families were part of a larger group of 20 kindreds thought to have atypical dementia or atypical Alzheimer's disease based on clinical and/or neuropathological findings (such as early-onset psychosis or predominant neurofibrillary tangles). When our group (Bird et al., 1997) linked the disease in the Seattle A (BK) family to 17q and then found a mutation in the tau gene in that family (Poorkaj et al., 1998), we began systematically evaluating our other atypical dementia families for mutations in tau. We found a P301L mutation in the Seattle D family (Poorkaj et al., 1998) simultaneously with the discovery of several additional mutations in FTDP-17 kindreds by Hutton et al. (1998) and Spillantini et al. (1998b). We have now identified three families from our study population with the P301L tau mutation, and they form the basis for the present report. All living family members were evaluated and examined by one of the authors whenever possible. Most of the living affected persons are mute and unable to follow commands, making formal testing difficult and incomplete. Most affected members of these families are deceased and medical records and neuroimaging studies on these persons were obtained and reviewed. Spouses and unaffected family members were also interviewed. Age of onset was determined to be that age at which the subject first demonstrated behavioural or personality change or memory loss, as noted by family, friends and colleagues. Blood samples were obtained from living family members with informed consent approved by the institutional Human Subjects Review Boards.
Brain tissue was obtained at autopsy from three affected people in two families (Families D and F). Brain tissue was preserved in formalin and slides were prepared from paraffin-embedded blocks. No frozen tissue was available. Sections were stained with haematoxylin and eosin, Bielschowsky stain and thioflavin S, and immunostained with antibodies to tau 2 (1 : 12 000; Sigma, St Louis, Mo., USA), PHF1 (1 : 800; Sharon Greenberg) and β amyloid1–28 (10D5, 1 : 5000; Athena Neuroscience, Worchester, Mass., USA).
DNA was extracted from peripheral blood leucocytes and the tau gene was sequenced by previously reported methods (Clark et al., 1998; Poorkaj et al., 1998). Apo E genotyping was performed by the restriction digest method of Hixson and Vernier (1994).
Results
Family D
The pedigree of Family D is shown in Fig. 1A. Mean age of onset in this family is 49.0 ± 5.1 years, mean disease duration is 5.7 ± 0.5 years and mean age at death is 56.3 ± 6.4 years (Table 1). Typical characteristics of the disease in this family are demonstrated by the index case (III-4). He had a masters degree in education and was a successful high school vocational education teacher. His difficulties began at age 48 years, when he was forced to retire after an alleged incident of threatening a student. He was unable to find further employment. Personal hygiene declined, his home was foreclosed, he was unable to handle his finances and he began living in a motel or his car. He spent most of his time walking the streets and was often arrested for loitering. At age 51 years he was brought to a hospital by the police who found him confused, talking to himself and having pulled a knife on an adolescent boy. He was disoriented to time, had disorganized and grandiose thoughts, was emotionally labile and sexually inappropriate. He was involuntarily hospitalized and then moved to a group home followed by a nursing home. At age 52 years his dementia had progressed and included memory problems, language difficulties, perseveration, agitation and aggressive, irritable behaviour. Neuropsychological evaluation revealed severely impaired attention, concentration, judgement and problem-solving (Table 2). The patient scored 9/30 on the Mini-Mental State Examination (MMSE) and 73/144 on the Mattis Dementia Rating Scale. The scores are in the `severely impaired' range for both screening measures. Although the patient was able to comprehend and follow directions, he had difficulty sustaining attention and putting forth effort without encouragement from the examiner. Word-finding difficulties and cognitive perseverations were evident. Despite these severe deficits, the patient was able to draw simple figures with minimal difficulty and simple attention was also intact. Although his performance was slow, he was able to complete a task of sustained simple attention, the Trail Making Test Part A. At age 56 years he was fully awake and alert but almost completely mute. He would occasionally say `yes' or `no' and rarely uttered a two- or three-word sentence. He was ambulatory and frequently paced for many hours. He demonstrated hyperorality and hyperphagia. He did not have rigidity, tremor, myoclonus or pathological reflexes. He would sometimes follow simple commands, but his overall performance was characterized by lack of motivation, lack of initiative and extreme response latencies. He did not appear to read, but could accurately perform tasks such as sorting a deck of cards by number. He consistently recognized his sister. He was unable to complete the Trail Making Test Part A. MMSE could not be formally tested but his score was likely to be lower than the 9/30 score he obtained at age 52 years. Utilization behaviour was also evident. Due to these limitations, he had testing which minimized or did not require a verbal response.
He was able to copy the complex Rey–Osterrieth figure and his performance was within the normal range, although the time he took to copy the figure was unusually long and he frequently stopped and put his pencil down (Fig. 2A). It was necessary for the examiner to prompt and encourage the patient to complete the task, in essence providing the continuous initiation and effort the patient was lacking. To further explore possible discrepancies between constructional tasks that have an executive function component (e.g. planning, initiation, execution and self-monitoring) and those that rely more exclusively on occipital/parietal abilities (e.g. perception and visual matching), the patient was given a `draw to command' and `draw to copy' task. In this test, the patient is asked first to draw an object (e.g. a flower) on a blank piece of paper. Next, the patient is given a flower to copy. In this manner the command and copy conditions can be compared. In three out of four commands, the patient was unable to produce the correct drawing. In contrast, he was easily able to correctly copy all four drawings (Fig. 2B). These results highlight the difference between a constructional task requiring executive function abilities (drawing to command), mediated by the frontal lobes, and a task that requires only constructional and visual–perceptual abilities (copying), mediated by the parietal and occipital lobes.
A CT brain scan at age 52 years and an MRI brain scan at age 56 years revealed severe bilateral frontal and temporal atrophy, the right side being worse than the left (Fig. 3A). CSF tau concentration was in the normal range (248 pg/ml; Athena Diagnostics, Worchester, Mass., USA).
Subject I-1 was the maternal grandfather of patient III-4 and worked as a ship's captain on the Great Lakes. He retired in his early fifties for unknown reasons. Following retirement he lived at home and his wife took complete care of him. He could not be left alone. His granddaughter recalls him continually paging through magazines without apparent comprehension. To keep him occupied his wife would give him a book and pencil and he would colour in all the Os on every page. He died in his sixties.
Subject II-2 was a nurse. At age 50 years she developed a personality change, became agitated and argumentative, stopped doing household chores and was caught shoplifting from a store. At age 52 years she was fired from her job for falsifying records and not showing up for work. Initially her family did not notice speech or memory problems. She began to eat constantly and gained 50 pounds. She would eat inappropriate items and once drank a quart of cooking oil. She roamed the house at night and cooked at night, often burning food and once burning herself. She rummaged through drawers and cabinets. Her personal hygiene deteriorated and she refused to bathe or change her clothes. Her attention span decreased and she could only follow simple commands. She spent much of her time colouring in colouring books. She developed word-finding problems, gradually spoke less, her speech became a babble and she could finally say only `yeah'. Her balance was noted to be poor but she continued to walk. She was admitted to a nursing home at age 65 years and died at age 66 years.
Subject II-3 was a mechanical engineer who noted the onset of left-hand tremor at age 41 years. This became progressively worse and spread to his right side. Serial examinations between ages 42 and 44 years showed progression of the physical findings. His mental status remained normal and he had no language deficits other than difficulties with articulation. His initial examination showed both resting and intention tremor of the left hand with mild cogwheel rigidity. His face was slightly expressionless and his speech was of low amplitude and high pitch. His tendon reflexes were hyperactive and symmetrical with downgoing plantar reflexes. He developed increasing rigidity that was eventually described as severe, as well as resting and intention tremor, all consistently worse on the left side. Diffuse hyper-reflexia and ankle clonus continued. EEG and lumbar puncture with CSF protein and cell count were normal. A CT brain scan showed slight prominence of the sulci over the cerebral convexity. An EMG showed a few positive waves and fasciculations in the first dorsal interosseii bilaterally. There was initially a mild improvement with l-dopa treatment, but he quickly became resistant to increasingly high doses. His diagnosis was atypical Parkinson's disease or striatonigral degeneration. At age 44 years he underwent stereotaxic surgery with a lesion in the right ventral lateral nucleus of the thalamus and the medial globus pallidus. Postoperatively there was a decrease in the tremor on the left side but no improvement in his rigidity. He was completely refractory to l-dopa and the rigidity became so diffuse and severe that he was bedridden. He was unable to speak for the last 3 months of his life. However, his family remained convinced that his memory and thinking remained good and he did not become demented. His eye movements were described as normal up to age 44 years and were not described thereafter. He died at age 46 years. Neuropathological findings are described below.
Subject II-4 was a surgeon. At age 56 years his wife noted that he had word-finding problems, made up new words and had poor articulation. He became forgetful of recent events, his reading became less coherent and his writing became illegible. He had several automobile accidents. He became paranoid, critical of others, sullen and had mood swings. At age 59 years his surgical practice was restricted because of difficulties with technique. He divorced, moved to a new city and was unable to obtain a licence to practice. At age 60 years he appeared in an emergency room requesting treatment for knee pain. He was confused, agitated, angry, suspicious, dishevelled and dirty. He became combative and violent and was hospitalized at a mental health facility. He was found to be uncooperative and hyperkinetic. He would lie down, then stand up and pace, then lie down again. He refused to acknowledge any problems. He could not give a coherent history and appeared to confabulate. He had great difficulty reading, would mumble and could not name individual words. He was partly oriented but short-term memory and attention were poor. He denied auditory or visual hallucinations. His speech was random, rapid and the words and phrases were not consistent with his level of education. His thoughts appeared to be racing and tangential and showed flight of ideas. No abnormalities were noted on a brief physical examination. A CT brain scan demonstrated bilateral frontal and temporal atrophy. His diagnosis was possible Pick's disease. He was transferred to a nursing home and treated with chlorpromazine, buspirone, haloperidol and paroxetine. He did not improve and these medications were discontinued. At age 61 years he was unable to walk and lay in bed with his neck flexed off the pillow. He had a worried facial expression but could not respond verbally to any questions. He was able to carry out simple commands slowly. No abnormalities of cranial nerves were noted. He had severe, generalized rigidity with hyperactive and symmetrical tendon reflexes. Plantar reflexes were down. There was a mild postural tremor of both upper extremities. He was receiving vitamin B12 injections. He died at age 63 years. Neuropathological findings are described below.
Individual III-2 was a dental hygienist who stopped working at age 47 years. At about this age her husband referred to her entering a `possessive stage'. By this he meant she hid things around the house, took articles out of the neighbour's garage, put groceries in her pockets instead of the grocery cart and was arrested several times for shoplifting. She cut off all the lower branches of the trees in her garden. Her personal hygiene deteriorated and she developed uncontrollable eating, with a weight gain from 105 to 162 pounds. Her husband put a lock on the refrigerator and her weight gradually decreased. She had no initial problems with memory or speech. She stopped reading and writing in her early fifties. At age 51 years she was seen by a neurologist who found a normal neurological examination except for inappropriate behaviour and a profound change in personality. He was aware of her family history and thought that she probably had Alzheimer's disease. She had a normal EEG and an MRI brain scan was described as showing diffuse cerebrocortical atrophy. An I-123 cerebral perfusion study demonstrated decreased perfusion in the frontal and temporal–parietal regions bilaterally. She would roam the house at night, wander away, get lost and have to be returned by the police. She gradually spoke less and less and was mute by age 59 years. At present, at age 61 years, she sits in a chair and stares ahead, apparently recognizing no-one. Television does not hold her attention and she does not respond to any verbal commands. She occasionally turns the pages of a colouring book repetitively. She walks slowly with difficulty while someone holds both hands and pulls her forward. She is incontinent and requires full care for bathing, dressing and feeding.
The clinical characteristics of other family members are shown in Table 1. Family members tend to be highly educated professionals and initially successful in their careers. Antisocial behaviour, disinhibition, language deficits and excessive eating occurred in most affected persons, usually followed by severe dementia after a period of 5 years. Family member II-3 had a diagnosis of `atypical' Parkinson's disease that included tremor, rigidity and bradykinesia. He was atypical because of prominent long motor tract signs and lack of response to l-dopa. The tremor, but not other features, responded to a combined thalamotomy/pallidotomy at another institution. In the last 6 months of his life he became bedridden and mute, but continued to follow commands and his family did not consider him demented.
Neuropathology
Brain autopsy was performed on family member II-4, who died at age 63 years. The brain weighed 1078 g. Gross brain examination showed severe atrophy of the frontal lobes and moderate atrophy of the temporal lobes, but no atrophy of the amygdala or hippocampus. Histologically, however, there were neuronal loss and gliosis only in the temporal lobe and orbitofrontal cortex. The main feature in this case was the presence of classic flame and globose-type (intracellular or ghost) and unusual perinuclear (crescent and ring-like) neurofibrillary tangles (Fig. 4). Neurofibrillary tangles were found frequently in the frontal and temporal cortices, amygdala, substantia innominata, raphe, locus coeruleus and substantia nigra. They were seen only occasionally in the parietal and occipital lobes. Immunocytochemical stains showed the neurofibrillary tangles to be positive for tau 2 and PHF1 (paired helical filament). By electron microscopy the neurofibrillary tangles were composed of both straight and twisted filaments (Fig. 5). The twisted filaments had a periodicity of ~110 nm and a diameter at the constriction of ~32 nm. There was mild tau 2 staining (PHF1-negative) of the cytoplasm of microglial cells in the putamen and cortical white matter. There were no neuritic or diffuse plaques typical of Alzheimer's disease and anti-beta amyloid protein antibody stain was negative. There were also no ballooned neurons, Pick bodies or Lewy bodies.
An autopsy was also performed on family member II-3 in 1976 at another institution. The brain weighed 1430 g and no gross atrophy was noted. Slides from that autopsy were available for review. The substantia nigra showed marked neuronal loss, gliosis and macrophage infiltration. Many neurons with globose or flame-shaped neurofibrillary tangles were seen in the substantia nigra, caudate, putamen, thalamus, subthalamus, red nucleus, periaqueductal grey, hippocampus, presubiculum and parahippocampus. There were also silver- and/or tau 2-positive cytoplasmic glial inclusions in many of these areas. There were no neuritic plaques, diffuse plaques, Lewy bodies or Pick bodies. The neuropathological changes were most similar to those of progressive supranuclear palsy.
Family F
The pedigree of Family F is shown in Fig. 1B. Mean age of onset in this family is 61.2 ± 3.8 years, mean disease duration 8.0 ± 2.9 years and mean age at death 68.3 ± 6.7 years (Table 3). Note that there is a strong tendency for the mean age of onset to be later and the disease duration to be longer for affected members of Family F compared with those of Family D.
Individual II-1 was born in 1892 in a rural New England community. She had a grade school education, married at age 20 years and had 12 children. At the age of approximately 57 years she became socially withdrawn and confused. Her memory for recent events was poor and she would wander off, becoming lost. She began talking to people who were not there and attempted to iron clothes that were frozen out of doors on a clothes-line. Her confusion progressed and she was admitted to a state mental hospital at age 62 years. Except for her mental status examination, her neurological examination was described as normal, including cranial nerves, tendon and plantar reflexes, motor strength, sensation and speech articulation. She had no tremors. She was generally quiet, co-operative and cheerful. She was disoriented for place and time and had memory impairment for recent and remote events. She could speak in full, albeit inappropriate, sentences but had naming and word-finding difficulties. She could not name any of several simple objects shown to her nor could she choose their right name from a list. She could not give her own name but she was able to write her name with a pen. When asked to write the names of other objects she perseverated in writing her own name. Her condition deteriorated over the subsequent year. She became essentially mute (only using the word `yeah') and would constantly burst into spontaneous giggles. She had bowel and bladder incontinence. Her diagnosis was chronic brain syndrome with cerebral arteriosclerosis. She died in 1957 at age 65 years and an autopsy revealed metastatic renal cell carcinoma. The brain weighed 1100 g and was described as showing moderate thinning of the gyri and widening of the sulci. Only a single haematoxylin/eosin microscopic section of the basal ganglia was performed, and was described as normal (the slide was later discarded).
Individual III-8 had an eighth-grade education and was a self-taught, successful electrician. In his mid-fifties he developed a change in personality. He became rude to friends, lost interest in personal hygiene, lost memory for words and events, and `couldn't be reasoned with any more'. Examination at age 61 years revealed that he was alert and co-operative, without pathological reflexes. He had a mouth tic and a `nervous laugh'. He was easily confused, had word-finding difficulties and scored 21 out of 30 on the MMSE (Table 2). Language performance was severely impaired and he missed 7 out of the first 20 items of the Boston Naming Test (very simple words). There was a 21-point difference between verbal IQ (71) and performance IQ (92). He performed in the normal range on tasks involving manipulation of visual information and design characteristics, but had trouble reproducing drawings after a delay. MRI scan revealed severe bilateral frontotemporal atrophy (Fig. 3B). Behavioural and cognitive deterioration continued. He would run a riding mower in strange patterns, compulsively saved objects such as soda cans, and would consume entire bags of cookies at one sitting. Speech became hesitant and non-fluent, and he completely stopped talking at age 65 years. On examination at age 67 years he was awake and alert but mute. He spent most of the day pacing and walking in circles. His gait was normal and he had no parkinsonian features other than a mild masked face and positive glabellar reflex. He followed no commands but could be guided in some tasks such as sitting and lying. His upper and lower limbs were strong, without atrophy or fasciculations. He had bilateral diffuse paratonia. There was no tremor and no myoclonus. Deep tendon reflexes were bilaterally symmetrical and hyperactive. He had a snout reflex and brisk jaw-jerk and made sucking noises with his mouth.
Individual III-17 in Family F was a machinist with adult-onset diabetes mellitus. He complained of feeling despondent at age 61 years and was evaluated for increasing anxiety at age 62 years. No cause for his feelings could be determined and his physical examinations were normal. At age 66–67 years his family noted a definite personality change and he began `acting odd'. He made mistakes in his job and lost interest in his daily activities. He initially had a normal physical examination and unremarkable memory testing. However, a few months later he could not recall recent events and was unable to recall any of three names after 3 min. A CT scan demonstrated mild to moderate diffuse cortical atrophy and he was thought to have probable Alzheimer's disease. By age 68–69 years he had shown considerable deterioration and was incontinent. He was noted to have a very unsteady gait but no ataxia on finger-to-nose testing. He was almost mute and spoke only occasional words. He did not know his name, the date, the time or the place. He had poor upgaze and a masked face. Tendon reflexes were described as symmetrical and unremarkable. A repeat CT brain scan demonstrated frontal and temporal atrophy. By age 69 years he required assistance with all activities of daily living and at age 70 years he was placed in a chronic care nursing facility. He died at age 73 years. Neuropathological findings are described below.
Patient III-24 was a self-employed carpenter who was unable to complete his work at about age 61–62 years. For example, he became unable to read a ruler. His wife noted problems with his memory. He had a marked personality change with angry outbursts, secretive behaviour and accused his friends of stealing from him. He hit his wife on one occasion. A neurological examination found no difficulties with his co-operation, language or brief memory testing. His gait, cranial nerves, strength, muscle tone, tendon reflexes, plantar reflexes, coordination and sensory examination were reported as normal. He had no Parkinsonian features. MMSE was 23/30. At age 63 years a CT brain scan showed marked diffuse cortical atrophy most pronounced in the frontal and temporal regions. A technetium-99 SPECT (single photon emission computed tomography) brain-imaging study showed marked decreased activity bilaterally in the frontal lobes as well as in the right temporal region (Fig. 3C). CSF cell count, protein, glucose and VDRL (Venereal Disease Research Laboratory) test were normal. An extensive battery of laboratory tests was also normal, including blood glucose, electrolytes, blood urea nitrogen, calcium, alkaline phosphatase, complete blood count, thyroid stimulating hormone and serum vitamin B12. An EEG was regarded as minimally abnormal with excessive beta activity. The patient was treated with both tacrine and aricept without noticeable benefit. He had a period of hyperphagia with considerable weight gain. At age 66 years he is described as passive, very bradykinetic and mute. He requires assistance with dressing and bathing and is in a full-time nursing care facility.
The clinical characteristics of the other members of Family F are shown in Table 3. Personality change and language deficits are common and hyperphagia with weight gain was seen in four persons.
Neuropathology
The brain, spinal cord, skeletal muscle and peripheral nerve were obtained at autopsy from family member III-17, who died at the age of 73 years of bronchopneumonia. The brain weighed 1110 g. The right half of the brain was serially sectioned and snap-frozen. The left half of the brain was sectioned following fixation and sampled for histological evaluation. At the gross level there was circumscribed, knife-blade-like atrophy affecting the frontal and temporal lobes with sparing of the superior temporal gyri and the parietal and occipital lobes. The amygdalae and hippocampi were mildly atrophic, the substantia nigra was mildly pale and the locus coeruleus was not discernible. Microscopically there was severe loss of neurons and striking gliosis of the frontal, temporal, insular and entorhinal cortices and the amygdala. The hippocampus and subiculum were mildly gliotic but otherwise spared. Numerous ballooned neurons, with their nucleus and lipofuscin displaced peripherally by distended, eosinophilic, weakly to moderately argyrophilic material, were noted in neocortical regions and in the entorhinal cortex (Fig. 6). Occasional ballooned neurons contained peripheral cytoplasmic vacuoles. There was marked loss of pars compacta neurons in the substantia nigra. The caudate nucleus was small and mildly gliotic. There was mild gliosis of the anterior and dorsomedial thalamic nuclei and the medial subthalamic nucleus but all other nuclei were normal. Neither Pick bodies nor Lewy bodies were identified. Scattered neuritic plaques were noted in the neocortex, but they were insufficient in number to make a diagnosis of Alzheimer's disease using the criteria of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) (Mirra et al., 1991). Scant neurofibrillary tangles and neuritic plaques were noted in the hippocampus, subiculum and entorhinal cortex. Mixed neurogenic and disuse atrophy was present in the psoas and quadriceps. Peripheral nerves in this patient with a long history of adult-onset diabetes showed mild fibrosis but a normal complement of large- and medium-sized myelinated fibres and minimal vascular abnormality. Histological examination of multiple levels of the spinal cord and nerve roots revealed no abnormality.
Family G
The pedigree of Family G is shown in Fig. 1C. The mean age of onset of dementia in this family is 64.3 ± 6.7 years and the mean disease duration is 7.3 ± 1.7 years (Table 4). The index case (II-8) had an illness similar to that of the other family members. He had an eighth-grade education and worked as a manager in a café. His dementia was first noticed at the age of 61 years, when he became easily distracted, had poor concentration and attention and had difficulty calculating change with money. His thinking was concrete and showed poor insight. Neuropsychological evaluation revealed difficulties with language expression and comprehension (Table 2). Delayed recall performance, as tested by word list recall, was within the normal range, whereas his performance in the Wechsler Memory Scale—Revised paragraph story recall test was mildly impaired. His MMSE score was 26/30 and his Clinical Dementia Rating Scale (CDR) score was 1.0. There were no abnormalities on the non-cognitive portion of the neurological examination, including absence of parkinsonian signs and myoclonus. An MRI scan of the brain showed predominant frontotemporal atrophy, with both sulcal widening and severe enlargement of the frontal horns. At age 62 years he was no longer able to operate the register at the café. He was increasingly stubborn concerning shaving and bathing, although he was able to perform these activities independently. He was obsessed with food, frequently demanded meals and gained weight. He set fire to the kitchen while preparing a meal. His MMSE was 20/30. Over the next year the patient declined noticeably, both functionally and cognitively. He tended to wander and be up most of the night. He had an apparent fear of water. Frequent urinary incontinence and occasional bowel incontinence developed. He was still capable of recalling the names of relatives and old friends. At age 63 years his MMSE was 12/30 and he was now graded a CDR stage of 2.0. By age 64 years he was severely demented, requiring assistance for dressing, bathing and toileting. He could repeat no words, but could name a pencil and a watch. His MMSE score was 5/30 and his CDR was now 3.0. At age 65 years he developed frequent rhythmic habits such as tapping or slapping his knee. He was completely echolalic, with very little spontaneous speech. Moderate bradykinesia and paratonia were present. He stood with assistance and walked with a flexed posture and slightly wide base. Stride length was normal. There were prominent grasp, snout, and glabellar tap reflexes. There was no tremor or myoclonus. He scored 22/100 on the Severe Impairment Battery (100 being least impaired). At age 66 years the patient was admitted to a nursing home because of significant care demands. He currently has no spontaneous speech and does not recognize his wife or friends. He has limited mobility, requiring full assistance for eating, ambulation and transfers. He appears moderately parkinsonian, but does not have tremor or myoclonus.
Details of the dementia in other family members are presented in Table 4. One sister (II-5) presented at age 64 years with delusions and was initially given the diagnosis of paranoid schizophrenia, but progressed to a dementia syndrome with impairments in multiple cognitive domains including memory and language function. A second sister (II-3) was reported to have died of an identical dementia syndrome characterized by poor insight and judgement, and progressing rapidly to impairment in memory, language and problem-solving. There has been no autopsy with neuropathology in this family.
Neuropsychological test results
Results of detailed testing were available for one member of each family (Table 2). Despite differences in duration of illness and large differences in level of education between patients, similarities between the testing patterns remain evident. Overall, the patients demonstrated significant impairments in nearly all cognitive domains. The most notable exception, however, was the relative preservation of constructional abilities, which remained intact even to very late stages of the illness. For example, patient III-4 of Family D was able to copy the Rey–Osterrieth Complex Figure when he was essentially mute with the MMSE score of 9 (obtained 2 years prior to administration of this test) (Fig. 2). This finding is also reflected in excellent performance by two of the patients on the WAIS-R (Wisconsin Adult Intelligence Scale—Revised) Block Design subtest. The WAIS-R Block Design subtest is a fairly demanding task in which the patient must construct complex designs from two-dimensional stimuli using coloured blocks. The test is also designed such that the items are progressively more difficult. Therefore, our finding of preserved constructional ability is supported by tasks that are much more difficult than the simple drawing tasks such as those included in the MMSE, CERAD and Dementia Rating Scale. This finding of relatively preserved constructional abilities in frontotemporal dementia and Pick's disease patients has been reported previously (Mendez et al., 1996; Pachana et al., 1996; Cherrier et al., 1997).
The other area of cognitive ability that appears to be preserved for two of the patients (II-8 of Family G and II-8 of Family F) is memory. While both of these patients demonstrate impaired to mildly impaired verbal memory, their visual memory is intact. Intact visual memory and constructional abilities may be attributed to the relative preservation of the parietal lobe functions that underlie spatial abilities. SPECT studies in frontotemporal dementia reveal relative sparing of parietal and occipital regions, with the bifrontal and bitemporal areas demonstrating hypoperfusion in the early stages (Risberg et al., 1993; Read et al., 1995). Patient III-4 (Family D) did not demonstrate relatively preserved visual memory at the time of testing. However, this may have been due to the overall severity of his deficits at that time. Pachana et al. (1996) also found relatively preserved memory in a group of patients with frontotemporal dementia compared with Alzheimer's disease patients, particularly for recognition memory. Our results also demonstrate preserved recognition memory of both verbal and visual information. While recognition tasks are easier than free recall, the preservation of recognition memory suggests that information is being encoded and stored and can be accessed. This pattern is in contrast to Alzheimer's disease patients, who generally do not benefit from recognition tasks or who generate high rates of false-positive identifications (Mendez et al., 1996). In addition to intact memory, free recall of information also requires some degree of organization mediated by the frontal lobes, which could contribute to the difference between free recall and recognition in these two patients.
Serial MMSE evaluations were available on patient III-8 (Family G). Three years after the onset of symptoms, the patient dropped six points to a score of 20/30. Over the next year his score dropped two more points, followed by a drop of six points 3 months later and of seven points 7 months later. Overall, the patient dropped from a score of 26/30 at 2 years of illness to a score of 5/30 after 5 years of illness. Although age of onset is difficult to determine, these results suggest a rapid decline in cognitive abilities in the mid-stage of the illness.
In summary, the neuropsychological tests indicate relatively preserved constructional abilities, visual memory and recognition memory in these patients, with evidence of a relatively rapid decline in overall cognitive abilities over a 5-year period.
Analysis of tau gene
Sequencing of the tau gene revealed a C→T alteration at nucleotide 903 of exon 10 (Fig. 7 and diagram below).
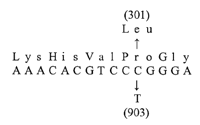
This represents a mutation in a highly conserved region of the second microtubule-binding domain, resulting in a leucine substitution for proline at codon 301 (P301L) (Locus Accession no. 2911249). This same mutation was identified in affected members of all three families and segregates with the disease (data not shown).
Apo E results
In the three families there were six members with the Apo E ε4 allele and seven without the ε4 allele (Tables 1–3). The mean ages of onset for these two groups were 58.8 ± 6.0 and 60.1 ± 6.4 years respectively; the difference is not significant (P > 0.10). Of the two persons with earliest onset (at ages 48 and 49 years) in Family D, one had the genotype ε3/3 (III-4) and one had the genotype ε3/4 (III-2). The person with latest onset of 67 years had the genotype ε3/4 (III-3, Family F).
Genealogy note
Each of these three families are of English and French-Canadian ancestry. It has not been possible to determine the exact ethnic background of the affected persons in the earliest generations. There is another French-Canadian family from Montreal with the P301L mutation (Clark et al., 1998). None of these four kindreds at present have any known family members in common (D. Geshwind, personal communication). It remains possible that two or more of these families have a common founding ancestor who carried the P301L tau mutation.
Discussion
The three families presented here represent a genetic form of frontotemporal dementia. The disease is inherited as an autosomal dominant trait in each family. Clinically the disease is characterized by behavioural changes, disinhibition, language deficits and eventual muteness. Visual memory abilities appear to be intact early in the disease but deteriorate as the disease progresses. Frontal executive functions, such as drawing to command, were more impaired than parietal–occipital functions requiring only visual–perceptual and construction abilities, such as copying a diagram, a common phenomenon seen in patients with frontotemporal dementia (Mendez et al., 1996; Snowden et al., 1996). Lobar atrophy of the frontal and temporal lobes is demonstrated by neuroimaging studies and gross brain pathology. Members of these three families usually did not have prominent Parkinsonian features early in the disease (except for patient II-3 of Family D). The kindreds are similar to several other previously reported families with variants of frontotemporal dementia, including the Seattle A family (BK) (Sumi et al., 1992; Bird et al., 1997), a family with multiple system tauopathy with presenile dementia (MSTD) (Spillantini et al., 1997, 1998a) and others reviewed by Foster et al. (1997).
All three families reported here have an identical missense mutation in the tau gene involving exon 10 and producing an amino acid change in the second microtubule-binding domain (P301L). This may interfere with the ability of tau to bind with microtubules and lead to intracellular filament formation and aggregation (Delacourte et al., 1998; Spillantini et al., 1998b). This mutation will affect only four-repeat tau, because exon 10 is spliced out of three-repeat tau isoforms (Delacourte et al., 1998). There is now strong evidence that mutations in tau cause hereditary forms of frontotemporal dementia (Clark et al., 1998; Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998b). The correlation is especially powerful because of the presence of obvious tau pathology in many of the families. This is further confirmed by the severe neurofibrillary tangles found in Family D of the present study. Mutations have been and are continuing to be discovered in several different regions of the tau gene, and phenotype–genotype comparisons will need to be made, including the present kindreds as well as additional families, in order to determine the relationship of specific mutations to clinical and pathological phenomena. The molecular mechanism of disease pathogenesis may be different in families with missense mutations in exon 10 compared with others in which the mutations are in the adjacent intronic splice site [dementia–disinhibition–parkinsonism–amyotrophy complex (DDPAC) and MSTD] (Clark et al., 1998; Hutton et al., 1998; Spillantini et al., 1998b). The missense mutations may produce abnormal microtubular binding by tau, whereas the splice-site mutations may produce an excess of four-repeat tau containing exon 10.
Because the three families have the same mutation, it is not surprising that they have many similar phenotypic characteristics. Members of each family generally experienced early behavioural changes (especially disinhibition) and language deficits associated with marked frontal and/or temporal cortical atrophy with neuronal loss and gliosis. Involvement of the amygdala and substantia nigra was also commonly noted at autopsy.
In view of the identical tau mutations it is intriguing to find substantial differences between and within the three families. Although numbers of cases are relatively small, Family D (II-4) had a mean age of onset >10 years younger than those of Families F and G (49 versus 61–64 years). There was also a tendency for disease duration to be shorter in Family D (5.3 versus 7.3–8 years). Of all affected members in these families, only a single individual in Family D had marked parkinsonian features, but these were prominent enough to result in neurosurgical treatment. There were also pathological differences. One brain from Family D showed severe and fairly diffuse neurofibrillary tangle formation. The neurofibrillary tangles had a remarkable circular and perinuclear distribution within the cytoplasm of neurons. This brain showed no ballooned cells. In contrast, the brain from Family F showed only a few neurofibrillary tangles in the hippocampus, subiculum and entorhinal cortex, which was thought to be within the expected range for the patient's age (73 years). However, this patient had extensive ballooned neurons or Pick cells, a finding that was not present in the brain from II-4 in Family D and was consistent with the diagnosis of Pick's disease, type B (Dickson, 1998). Ballooned neurons have been seen in other FTDP-17 families (Heutink et al., 1997). The other neuropathological specimen from Family D was similar to progressive supranuclear palsy, another disorder with tau pathology showing genetic disequilibrium with a tau gene polymorphism (Conrad et al., 1997). When the three brains from these families were initially studied in isolation they were each given different diagnoses (Parkinson's disease, Pick's disease type B and neurofibrillary tangle degeneration). It was not at all obvious that they might have an identical genetic aetiology.
A Dutch family (HFTD1; hereditary frontotemporal dementia 1) reported by Heutink et al. (1997) has also been found to have the P301L mutation in tau (Hutton et al., 1998). That family has many similarities to the three kindreds reported here. The similarities include mean age of onset (50.4 years), disease duration (8 years), disinhibition, restlessness, hyperorality, reduced speech output and impaired executive skills. Family HFTD1 also had frontotemporal atrophy, degeneration of the substantia nigra and ballooned neurons. Differences compared with our three families were the absence of neurofibrillary tangles and immunohistochemical studies negative for tau in the Dutch family. Our families also show that the age of onset can vary from the 5th to the 8th decade. It is unlikely that the P301L mutation will be associated with a highly specific and distinct clinical phenotype because of the variability reported both within and between different FTDP-17 families (Lynch et al., 1994; Foster et al., 1997). The prominent and consistent parkinsonism in the PPND (pallidopontonigral degeneration) family with a different mutation in exon 10 (N279K) may be a relative exception to this trend (Clark et al., 1998). Even so, marked parkinsonian features occasionally appear in many of the other FTDP-17 kindreds (including II-3 in our Family D and others reviewed by Foster et al., 1997).
The clinical and pathological differences seen in these families strongly suggest that, in addition to the missense mutation in exon 10 of the tau gene, there are other environmental and/or genetic factors influencing the phenotype. The Apo E allele ε4 is known to influence the clinical course of Alzheimer's disease primarily by reducing the average age of onset (Roses, 1994). Apo E does not appear to be playing such a role in the three families reported here or in the Seattle A family (BK) with FTDP-17 (Tsuang et al., 1997). It will be important to continue the search for other environmental and genetic factors influencing the FTDP-17 phenotype because of their important implications for potential treatment and prevention strategies.
Note added in proof
Since submission of this manuscript several reports have appeared describing additional aspects of the P301L tau mutation in frontotemporal dementia (Dumanchin et al., 1998; Hasegawa et al., 1998; Rizzu et al., 1998; Spillantini et al., 1998c).
Clinical characteristics of Family D
Clinical characteristics of Family D
Neuropsychological test results
| Subject | |||
|---|---|---|---|
| III-4 (Family D) | III-8 (Family F) | II-8 (Family G) | |
| Age at onset/age at testing (years) | 48/52 | 56/60 | 61/63 |
| Years of education | 18 | 8 | 8 |
| MMSE total | 9/30 | 21/30¶ | 26/30 |
| Illness duration at testing | 4 years | 4 years | 2 years |
| Subject | |||
|---|---|---|---|
| III-4 (Family D) | III-8 (Family F) | II-8 (Family G) | |
| Age at onset/age at testing (years) | 48/52 | 56/60 | 61/63 |
| Years of education | 18 | 8 | 8 |
| MMSE total | 9/30 | 21/30¶ | 26/30 |
| Illness duration at testing | 4 years | 4 years | 2 years |
| Cognitive domains Tests administered | Raw score and impairment level | Raw score and impairment level | Raw score and impairment level |
|---|---|---|---|
| *Test or item was not administered; †Wechsler Memory Scale I (immediate only); ‡administered 2 years after other test scores shown in the table, when the patient was essentially mute; §Wechsler Memory Scale I Story A only; ¶administered 1 year after other tests shown in the table; #WORLD spelled backwards was administered instead of serial sevens. D/C = item discontinued due to confusion or inability to complete; WNL = within normal limits; Aphasia Screening Test = Aphasia Screening Test as part of the Halstead Reitan Neuropsychological Battery (Reitan, no date); Boston Naming Test = 60-item version of the Boston Naming Test (Kaplan et al., 1983); CERAD = Consortium to Establish a Registry for Alzheimer's Disease, Neuropsychological Battery (Welsh et al., 1994); DRS = Dementia Rating Scale (Mattis, 1988); MMSE = Mini-Mental Status Examination (Folstein et al., 1975); NCSE = Neurobehavioral Cognitive Status Exam (Group, 1988); Ravens Progressive Matrices: see Raven et al. (1976); ROCFT = Rey–Osterrieth Complex Figure Test (Osterrieth, 1944); Severe Impairment Battery: see Saxton et al. (1993); Test for Severe Impairment: see Albert and Cohen (1992); Trails A and B = Trail Making Test (Reitan, 1985). | |||
| Orientation | |||
| MMSE | 2/10 impaired | 9/10 WNL | 9/10 WNL |
| NCSE | 2/9 impaired | * | * |
| DRS | 5/9 impaired | * | * |
| Simple attention | |||
| Trails A: total time | 209 impaired | * | * |
| Digit span forward | 4 impaired | 4 impaired | 5 impaired |
| Construction | |||
| MMSE | 1/1 WNL | 1/1 WNL | 1/1 WNL |
| NCSE | 3/3 WNL | * | * |
| DRS | 6/6 WNL | * | * |
| ROCFT | 34/36 WNL‡ | * | * |
| WAIS-R Block Design | * | 24/51 WNL | 11/51 WNL |
| CERAD praxis | * | * | 9/11 WNL |
| Language | |||
| Boston Naming Test | 11/60 impaired | 25/60 impaired | * |
| Aphasia Screening Test | 37 impaired | * | * |
| CERAD Naming | * | * | 10/15 impaired |
| Memory | |||
| WMS-R LM I | 3/57 impaired† | 6/24 impaired§ | 12/50 mildly impaired |
| WMS-R VR I | 2/14 impaired† | 7/14 WNL | 31/41 WNL |
| WMS Paired Associates | * | 10/22 WNL | * |
| WMS-R LM II | *† | 3/24 impaired§ | 8/50 mildly impaired |
| WMS-R VR II | *† | * | 23/41 WNL |
| Memory recall MMSE | 0/3 impaired | 1/3 impaired | 2/3 impaired |
| CERAD Word List | * | * | 22/30 impaired |
| CERAD Word List Delay | * | * | 6/10 WNL |
| CERAD Recognition | * | * | 10/10 WNL |
| DRS Memory | * | * | * |
| DRS Recognition | 9/9 WNL | * | * |
| Executive functions | |||
| Trails B Total Time | D/C | 186 impaired | * |
| Digit Span Backward | 0 impaired | 3 impaired | 3 impaired |
| Letter or category fluency | 0 impaired | * | 6 impaired |
| NCSE Judgement | 1/6 impaired | * | * |
| NCSE Similarities | 0/8 impaired | * | * |
| DRS Similarities | 13/24 impaired | * | * |
| WAIS-R Similarities | * | 0/28 impaired | 0/28 impaired |
| Ravens Progressive Matrices | * | 24/60 WNL | * |
| Calculations | |||
| Serial Sevens MMSE | 0/5 impaired | *# | *# |
| Calculations NCSE | 0/4 impaired | * | * |
| WAIS-R Arithmetic | * | 6/19 mildly impaired | * |
| Severe impairment battery | * | * | 22/100 impaired |
| Test for severe impairment | 21/24 impaired | * | * |
| Cognitive domains Tests administered | Raw score and impairment level | Raw score and impairment level | Raw score and impairment level |
|---|---|---|---|
| *Test or item was not administered; †Wechsler Memory Scale I (immediate only); ‡administered 2 years after other test scores shown in the table, when the patient was essentially mute; §Wechsler Memory Scale I Story A only; ¶administered 1 year after other tests shown in the table; #WORLD spelled backwards was administered instead of serial sevens. D/C = item discontinued due to confusion or inability to complete; WNL = within normal limits; Aphasia Screening Test = Aphasia Screening Test as part of the Halstead Reitan Neuropsychological Battery (Reitan, no date); Boston Naming Test = 60-item version of the Boston Naming Test (Kaplan et al., 1983); CERAD = Consortium to Establish a Registry for Alzheimer's Disease, Neuropsychological Battery (Welsh et al., 1994); DRS = Dementia Rating Scale (Mattis, 1988); MMSE = Mini-Mental Status Examination (Folstein et al., 1975); NCSE = Neurobehavioral Cognitive Status Exam (Group, 1988); Ravens Progressive Matrices: see Raven et al. (1976); ROCFT = Rey–Osterrieth Complex Figure Test (Osterrieth, 1944); Severe Impairment Battery: see Saxton et al. (1993); Test for Severe Impairment: see Albert and Cohen (1992); Trails A and B = Trail Making Test (Reitan, 1985). | |||
| Orientation | |||
| MMSE | 2/10 impaired | 9/10 WNL | 9/10 WNL |
| NCSE | 2/9 impaired | * | * |
| DRS | 5/9 impaired | * | * |
| Simple attention | |||
| Trails A: total time | 209 impaired | * | * |
| Digit span forward | 4 impaired | 4 impaired | 5 impaired |
| Construction | |||
| MMSE | 1/1 WNL | 1/1 WNL | 1/1 WNL |
| NCSE | 3/3 WNL | * | * |
| DRS | 6/6 WNL | * | * |
| ROCFT | 34/36 WNL‡ | * | * |
| WAIS-R Block Design | * | 24/51 WNL | 11/51 WNL |
| CERAD praxis | * | * | 9/11 WNL |
| Language | |||
| Boston Naming Test | 11/60 impaired | 25/60 impaired | * |
| Aphasia Screening Test | 37 impaired | * | * |
| CERAD Naming | * | * | 10/15 impaired |
| Memory | |||
| WMS-R LM I | 3/57 impaired† | 6/24 impaired§ | 12/50 mildly impaired |
| WMS-R VR I | 2/14 impaired† | 7/14 WNL | 31/41 WNL |
| WMS Paired Associates | * | 10/22 WNL | * |
| WMS-R LM II | *† | 3/24 impaired§ | 8/50 mildly impaired |
| WMS-R VR II | *† | * | 23/41 WNL |
| Memory recall MMSE | 0/3 impaired | 1/3 impaired | 2/3 impaired |
| CERAD Word List | * | * | 22/30 impaired |
| CERAD Word List Delay | * | * | 6/10 WNL |
| CERAD Recognition | * | * | 10/10 WNL |
| DRS Memory | * | * | * |
| DRS Recognition | 9/9 WNL | * | * |
| Executive functions | |||
| Trails B Total Time | D/C | 186 impaired | * |
| Digit Span Backward | 0 impaired | 3 impaired | 3 impaired |
| Letter or category fluency | 0 impaired | * | 6 impaired |
| NCSE Judgement | 1/6 impaired | * | * |
| NCSE Similarities | 0/8 impaired | * | * |
| DRS Similarities | 13/24 impaired | * | * |
| WAIS-R Similarities | * | 0/28 impaired | 0/28 impaired |
| Ravens Progressive Matrices | * | 24/60 WNL | * |
| Calculations | |||
| Serial Sevens MMSE | 0/5 impaired | *# | *# |
| Calculations NCSE | 0/4 impaired | * | * |
| WAIS-R Arithmetic | * | 6/19 mildly impaired | * |
| Severe impairment battery | * | * | 22/100 impaired |
| Test for severe impairment | 21/24 impaired | * | * |
Neuropsychological test results
| Subject | |||
|---|---|---|---|
| III-4 (Family D) | III-8 (Family F) | II-8 (Family G) | |
| Age at onset/age at testing (years) | 48/52 | 56/60 | 61/63 |
| Years of education | 18 | 8 | 8 |
| MMSE total | 9/30 | 21/30¶ | 26/30 |
| Illness duration at testing | 4 years | 4 years | 2 years |
| Subject | |||
|---|---|---|---|
| III-4 (Family D) | III-8 (Family F) | II-8 (Family G) | |
| Age at onset/age at testing (years) | 48/52 | 56/60 | 61/63 |
| Years of education | 18 | 8 | 8 |
| MMSE total | 9/30 | 21/30¶ | 26/30 |
| Illness duration at testing | 4 years | 4 years | 2 years |
| Cognitive domains Tests administered | Raw score and impairment level | Raw score and impairment level | Raw score and impairment level |
|---|---|---|---|
| *Test or item was not administered; †Wechsler Memory Scale I (immediate only); ‡administered 2 years after other test scores shown in the table, when the patient was essentially mute; §Wechsler Memory Scale I Story A only; ¶administered 1 year after other tests shown in the table; #WORLD spelled backwards was administered instead of serial sevens. D/C = item discontinued due to confusion or inability to complete; WNL = within normal limits; Aphasia Screening Test = Aphasia Screening Test as part of the Halstead Reitan Neuropsychological Battery (Reitan, no date); Boston Naming Test = 60-item version of the Boston Naming Test (Kaplan et al., 1983); CERAD = Consortium to Establish a Registry for Alzheimer's Disease, Neuropsychological Battery (Welsh et al., 1994); DRS = Dementia Rating Scale (Mattis, 1988); MMSE = Mini-Mental Status Examination (Folstein et al., 1975); NCSE = Neurobehavioral Cognitive Status Exam (Group, 1988); Ravens Progressive Matrices: see Raven et al. (1976); ROCFT = Rey–Osterrieth Complex Figure Test (Osterrieth, 1944); Severe Impairment Battery: see Saxton et al. (1993); Test for Severe Impairment: see Albert and Cohen (1992); Trails A and B = Trail Making Test (Reitan, 1985). | |||
| Orientation | |||
| MMSE | 2/10 impaired | 9/10 WNL | 9/10 WNL |
| NCSE | 2/9 impaired | * | * |
| DRS | 5/9 impaired | * | * |
| Simple attention | |||
| Trails A: total time | 209 impaired | * | * |
| Digit span forward | 4 impaired | 4 impaired | 5 impaired |
| Construction | |||
| MMSE | 1/1 WNL | 1/1 WNL | 1/1 WNL |
| NCSE | 3/3 WNL | * | * |
| DRS | 6/6 WNL | * | * |
| ROCFT | 34/36 WNL‡ | * | * |
| WAIS-R Block Design | * | 24/51 WNL | 11/51 WNL |
| CERAD praxis | * | * | 9/11 WNL |
| Language | |||
| Boston Naming Test | 11/60 impaired | 25/60 impaired | * |
| Aphasia Screening Test | 37 impaired | * | * |
| CERAD Naming | * | * | 10/15 impaired |
| Memory | |||
| WMS-R LM I | 3/57 impaired† | 6/24 impaired§ | 12/50 mildly impaired |
| WMS-R VR I | 2/14 impaired† | 7/14 WNL | 31/41 WNL |
| WMS Paired Associates | * | 10/22 WNL | * |
| WMS-R LM II | *† | 3/24 impaired§ | 8/50 mildly impaired |
| WMS-R VR II | *† | * | 23/41 WNL |
| Memory recall MMSE | 0/3 impaired | 1/3 impaired | 2/3 impaired |
| CERAD Word List | * | * | 22/30 impaired |
| CERAD Word List Delay | * | * | 6/10 WNL |
| CERAD Recognition | * | * | 10/10 WNL |
| DRS Memory | * | * | * |
| DRS Recognition | 9/9 WNL | * | * |
| Executive functions | |||
| Trails B Total Time | D/C | 186 impaired | * |
| Digit Span Backward | 0 impaired | 3 impaired | 3 impaired |
| Letter or category fluency | 0 impaired | * | 6 impaired |
| NCSE Judgement | 1/6 impaired | * | * |
| NCSE Similarities | 0/8 impaired | * | * |
| DRS Similarities | 13/24 impaired | * | * |
| WAIS-R Similarities | * | 0/28 impaired | 0/28 impaired |
| Ravens Progressive Matrices | * | 24/60 WNL | * |
| Calculations | |||
| Serial Sevens MMSE | 0/5 impaired | *# | *# |
| Calculations NCSE | 0/4 impaired | * | * |
| WAIS-R Arithmetic | * | 6/19 mildly impaired | * |
| Severe impairment battery | * | * | 22/100 impaired |
| Test for severe impairment | 21/24 impaired | * | * |
| Cognitive domains Tests administered | Raw score and impairment level | Raw score and impairment level | Raw score and impairment level |
|---|---|---|---|
| *Test or item was not administered; †Wechsler Memory Scale I (immediate only); ‡administered 2 years after other test scores shown in the table, when the patient was essentially mute; §Wechsler Memory Scale I Story A only; ¶administered 1 year after other tests shown in the table; #WORLD spelled backwards was administered instead of serial sevens. D/C = item discontinued due to confusion or inability to complete; WNL = within normal limits; Aphasia Screening Test = Aphasia Screening Test as part of the Halstead Reitan Neuropsychological Battery (Reitan, no date); Boston Naming Test = 60-item version of the Boston Naming Test (Kaplan et al., 1983); CERAD = Consortium to Establish a Registry for Alzheimer's Disease, Neuropsychological Battery (Welsh et al., 1994); DRS = Dementia Rating Scale (Mattis, 1988); MMSE = Mini-Mental Status Examination (Folstein et al., 1975); NCSE = Neurobehavioral Cognitive Status Exam (Group, 1988); Ravens Progressive Matrices: see Raven et al. (1976); ROCFT = Rey–Osterrieth Complex Figure Test (Osterrieth, 1944); Severe Impairment Battery: see Saxton et al. (1993); Test for Severe Impairment: see Albert and Cohen (1992); Trails A and B = Trail Making Test (Reitan, 1985). | |||
| Orientation | |||
| MMSE | 2/10 impaired | 9/10 WNL | 9/10 WNL |
| NCSE | 2/9 impaired | * | * |
| DRS | 5/9 impaired | * | * |
| Simple attention | |||
| Trails A: total time | 209 impaired | * | * |
| Digit span forward | 4 impaired | 4 impaired | 5 impaired |
| Construction | |||
| MMSE | 1/1 WNL | 1/1 WNL | 1/1 WNL |
| NCSE | 3/3 WNL | * | * |
| DRS | 6/6 WNL | * | * |
| ROCFT | 34/36 WNL‡ | * | * |
| WAIS-R Block Design | * | 24/51 WNL | 11/51 WNL |
| CERAD praxis | * | * | 9/11 WNL |
| Language | |||
| Boston Naming Test | 11/60 impaired | 25/60 impaired | * |
| Aphasia Screening Test | 37 impaired | * | * |
| CERAD Naming | * | * | 10/15 impaired |
| Memory | |||
| WMS-R LM I | 3/57 impaired† | 6/24 impaired§ | 12/50 mildly impaired |
| WMS-R VR I | 2/14 impaired† | 7/14 WNL | 31/41 WNL |
| WMS Paired Associates | * | 10/22 WNL | * |
| WMS-R LM II | *† | 3/24 impaired§ | 8/50 mildly impaired |
| WMS-R VR II | *† | * | 23/41 WNL |
| Memory recall MMSE | 0/3 impaired | 1/3 impaired | 2/3 impaired |
| CERAD Word List | * | * | 22/30 impaired |
| CERAD Word List Delay | * | * | 6/10 WNL |
| CERAD Recognition | * | * | 10/10 WNL |
| DRS Memory | * | * | * |
| DRS Recognition | 9/9 WNL | * | * |
| Executive functions | |||
| Trails B Total Time | D/C | 186 impaired | * |
| Digit Span Backward | 0 impaired | 3 impaired | 3 impaired |
| Letter or category fluency | 0 impaired | * | 6 impaired |
| NCSE Judgement | 1/6 impaired | * | * |
| NCSE Similarities | 0/8 impaired | * | * |
| DRS Similarities | 13/24 impaired | * | * |
| WAIS-R Similarities | * | 0/28 impaired | 0/28 impaired |
| Ravens Progressive Matrices | * | 24/60 WNL | * |
| Calculations | |||
| Serial Sevens MMSE | 0/5 impaired | *# | *# |
| Calculations NCSE | 0/4 impaired | * | * |
| WAIS-R Arithmetic | * | 6/19 mildly impaired | * |
| Severe impairment battery | * | * | 22/100 impaired |
| Test for severe impairment | 21/24 impaired | * | * |
Clinical characteristics of Family F
Clinical characteristics of Family F
Clinical characteristics of Family G
| Subject | Sex | Age at onset (years) | Age at death (years) | Personality change | Memory loss | Language defect | Hyper-reflexia | Parkinson's features | Tremor | Hyperoral/weight loss | FT | atrophyApo E allele |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean age at onset 64.3 ± 6.7 years (n = 4); mean age at death 72.3 ± 5.4 years (n = 4); mean disease duration 7.3 ± 1.7 years (n = 3);u = unknown. | ||||||||||||
| I-1 | F | 75 | 80 | u | Yes | u | u | u | u | u | u | u |
| II-1 | M | u | 71 | u | Yes | u | u | u | u | u | u | u |
| II-3 | F | 57 | 65 | Yes | Yes | Yes | u | u | u | u | u | u |
| II-5 | F | 64 | 73 | Yes | Yes | Yes | u | u | u | u | u | 2/3 |
| II-8 | M | 61 | – | Yes | Yes | Yes | No | Yes | No | Yes | Yes | 2/4 |
| Subject | Sex | Age at onset (years) | Age at death (years) | Personality change | Memory loss | Language defect | Hyper-reflexia | Parkinson's features | Tremor | Hyperoral/weight loss | FT | atrophyApo E allele |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean age at onset 64.3 ± 6.7 years (n = 4); mean age at death 72.3 ± 5.4 years (n = 4); mean disease duration 7.3 ± 1.7 years (n = 3);u = unknown. | ||||||||||||
| I-1 | F | 75 | 80 | u | Yes | u | u | u | u | u | u | u |
| II-1 | M | u | 71 | u | Yes | u | u | u | u | u | u | u |
| II-3 | F | 57 | 65 | Yes | Yes | Yes | u | u | u | u | u | u |
| II-5 | F | 64 | 73 | Yes | Yes | Yes | u | u | u | u | u | 2/3 |
| II-8 | M | 61 | – | Yes | Yes | Yes | No | Yes | No | Yes | Yes | 2/4 |
Clinical characteristics of Family G
| Subject | Sex | Age at onset (years) | Age at death (years) | Personality change | Memory loss | Language defect | Hyper-reflexia | Parkinson's features | Tremor | Hyperoral/weight loss | FT | atrophyApo E allele |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean age at onset 64.3 ± 6.7 years (n = 4); mean age at death 72.3 ± 5.4 years (n = 4); mean disease duration 7.3 ± 1.7 years (n = 3);u = unknown. | ||||||||||||
| I-1 | F | 75 | 80 | u | Yes | u | u | u | u | u | u | u |
| II-1 | M | u | 71 | u | Yes | u | u | u | u | u | u | u |
| II-3 | F | 57 | 65 | Yes | Yes | Yes | u | u | u | u | u | u |
| II-5 | F | 64 | 73 | Yes | Yes | Yes | u | u | u | u | u | 2/3 |
| II-8 | M | 61 | – | Yes | Yes | Yes | No | Yes | No | Yes | Yes | 2/4 |
| Subject | Sex | Age at onset (years) | Age at death (years) | Personality change | Memory loss | Language defect | Hyper-reflexia | Parkinson's features | Tremor | Hyperoral/weight loss | FT | atrophyApo E allele |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean age at onset 64.3 ± 6.7 years (n = 4); mean age at death 72.3 ± 5.4 years (n = 4); mean disease duration 7.3 ± 1.7 years (n = 3);u = unknown. | ||||||||||||
| I-1 | F | 75 | 80 | u | Yes | u | u | u | u | u | u | u |
| II-1 | M | u | 71 | u | Yes | u | u | u | u | u | u | u |
| II-3 | F | 57 | 65 | Yes | Yes | Yes | u | u | u | u | u | u |
| II-5 | F | 64 | 73 | Yes | Yes | Yes | u | u | u | u | u | 2/3 |
| II-8 | M | 61 | – | Yes | Yes | Yes | No | Yes | No | Yes | Yes | 2/4 |
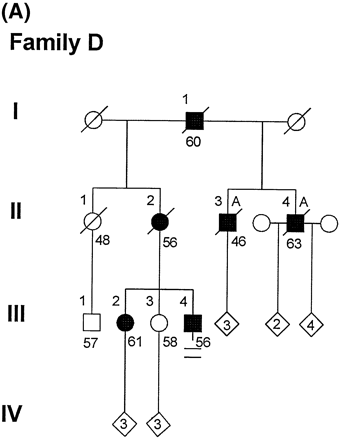
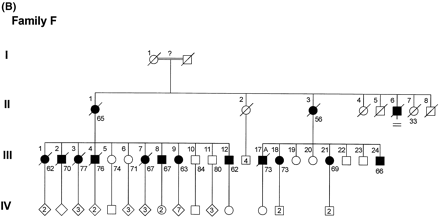
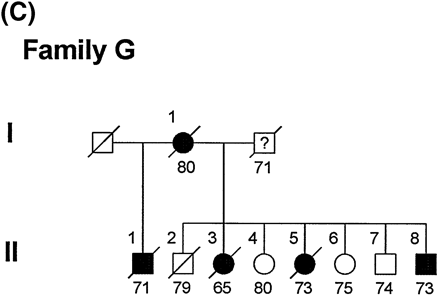
(A) Pedigree of Family D. Ages beneath each symbol are present age or age at death. Death is indicated by a diagonal line through the symbol. A = autopsy. (B) Pedigree of Family F. (C) Pedigree of Family G.
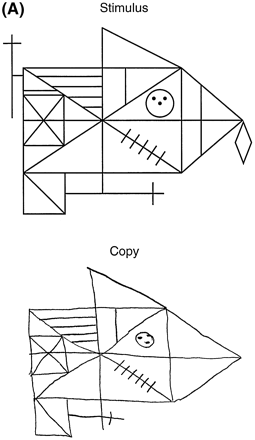
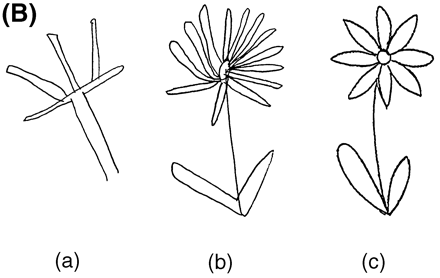
Essentially mute at age 56 years, subject III-4 in Family D was able to slowly make a good copy of the complex Rey–Osterrieth figure. (A) The test stimulus figure is at the top and the subject's copy is at the bottom. (B) The subject's attempt to draw a flower from memory (a) and his much better copy of a flower (b); (c) is the test stimulus flower. These drawings illustrate his superior ability at a constructional task (copying a figure) requiring only visuoperceptual abilities mediated by the parietal–occipital lobes compared with his poorer performance on a task (drawing to command) requiring frontal executive functions.
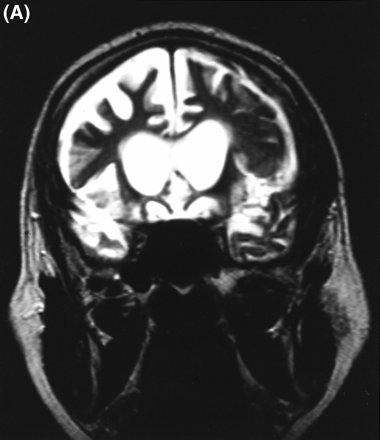
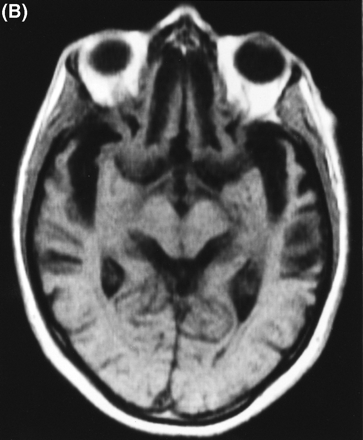
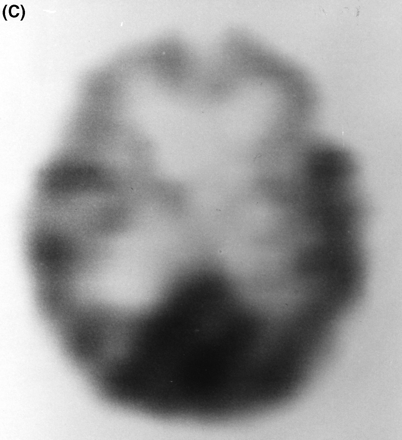
(A) T2-weighted MRI scan of subject III-4 from Family D at age 56 years, showing marked frontal atrophy, the right sidebeing worse than the left. (B) MRI scan of subject III-8 from Family F at age 61 years, showing severe bilateral temporal atrophy.(C) Technetium-99 (25.8 mCi) perfusion scan performed on subject III-24 in Family F at age 63 years. Decreased perfusion bilaterally in the frontal regions and in the right temporal region is demonstrated.
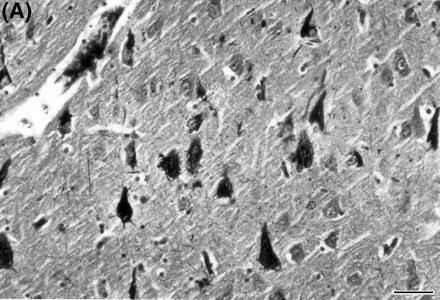
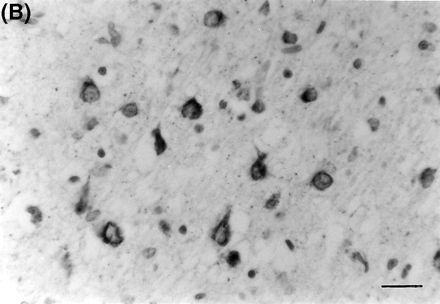
(A) Multiple neurons in the CA1 region of the hippocampus from subject II-4 of Family D, showingdense neurofibrillary tangle formation. Bielschowsky stain. Bar = 50 μm. (B) Neurons in the entorhinal cortex of the same subject, showing crescent- and ring-like perinuclear tangles. PHF1.Bar = 25 μm.
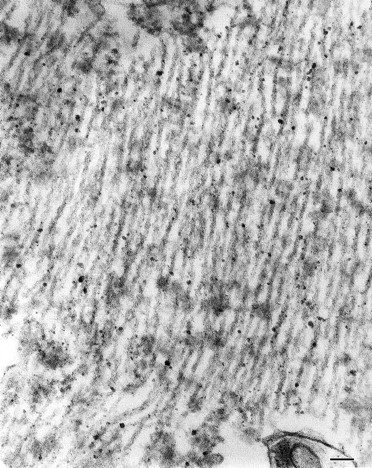
Electron micrograph of neuronal neurofibrillary tangles from parahippocampus of subject II-4 of Family D, showing twisted filaments with a periodicity of ~110 nm and a diameter of ~32 nm. Straight filaments were also seen. Bar = 200 nm.
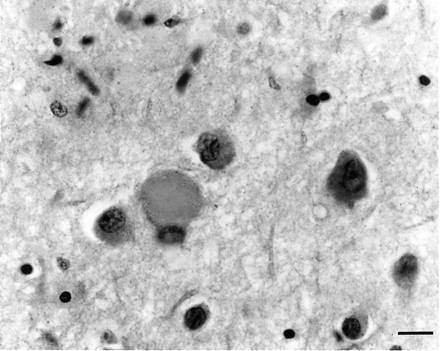
Ballooned neuron in the gliotic temporal lobe neocortex of subject III-17 from Family F. Luxol fast blue–haematoxylin. Bar = 50 μm.
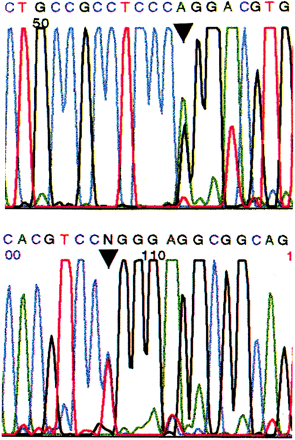
Applied Biosystems electropherogram DNA sequence pattern from subject III-4 in Family D showing the C→T alteration (arrowhead) at nucleotide 903 in exon 10 of the tau gene, sequenced with forward primer 10F (top) and sequenced with reverse primer 10R (bottom) (Poorkaj et al., 1998).
This work was supported by VA Medical Research funds and NIA Alzheimer Disease Research Center Grants AG 05136 and AG 08017. The Department of Pathology of University of Iowa provided autopsy material from II-3 in Family D. R. Fowler evaluated III-8 in Family F.
References
Albert M, Cohen C. The Test for Severe Impairment: an instrument for the assessment of patients with severe cognitive dysfunction.
Bird TD, Wijsman, E, Nochlin, D, Leehey M, Sumi SM, Payami H, et al. Chromosome 17 and hereditary dementia: linkage studies with three non-Alzheimer families and kindreds with late-onset FAD.
Cherrier MM, Mendez MF, Perryman K, Pachana NA, Miller BL, Cummings JL. Frontotemporal dementia versus vascular dementia: differential features on mental status examination.
Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17.
Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy.
Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y, et al. Vulnerable neuronal subsets in Alzheimer's and Pick's disease are distinguished by their tau isoform distribution and phosphorylation.
Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism.
Feany MB, Dickson DW. Neurodegenerative disorders with extensive tau pathology: a comparative study and review. [Review].
Folstein MF, Folstein SE, McHugh PR. `Mini-mental state': a practical method for grading the cognitive state of patients for the clinician.
Foster NL, Wilhelmsen K, Anders AF, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a consensus conference. [Review].
Group TNCN. Neurobehavioral cognitive status examination. Fairfax (CA): Northern California Neurobehavioral Group; 1998.
Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly.
Heutink P, Stevens M, Rizzu P, Bakker E, Kros JM, Tibben A, et al. Hereditary frontotemporal dementia is linked to chromosome 17q21–q22: a genetic and clinicopathological study of three Dutch families.
Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice site mutations in tau with the inherited dementia FTDP-17.
Lynch T, Sano M, Marder KS, Bell, KL, Foster NL, Defendini RF, et al. Clinical characteristics of a family with chromosome 17-linked disinhibition–dementia–parkinsonism–amyotrophy complex [see comments].
Mendez MF, Cherrier M, Perryman KM, Pachana N, Miller BL, Cummings JL, et al. Frontotemporal dementia versus Alzheimer's disease: differential cognitive features.
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease.
Osterrieth PA. Le test de copie d'une figure complexe: contribution a l'étude de la perception et de la memoire.
Pachana NA, Boone KB, Miller BL, Cummings JL, Berman N, et al. Comparison of neuropsychological and neurobehavioral functioning in Alzheimer's disease and frontotemporal dementia.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garutto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia.
Read SL, Miller BL, Mena I, Kim R, Itabashi H, Darby A, et al. SPECT in dementia: clinical and pathological correlation.
Reitan RM. Validity of the Trail Making Test as an indicator of organic brain damage.
Reitan RM. Instructions and procedures for administrating the psychological test battery used at the Neuropsychology Laboratory. Indianapolis (IN): Indiana University Medical Center; No date.
Risberg J, Passant U, Warkentin S, Gustafson L. Regional cerebral blood flow in frontal dementia of the non-Alzheimer type.
Rizzu P, van Swieten JC, Joossee M, Hasegawa M, Stevens M, Tibben A, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands.
Roses AD. Apolipoprotein E affects the rate of Alzheimer disease expression: β-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease [see comments]. [Review].
Saxton J, McGonigle KL, Swihart AA, et al. The Severe Impairment Battery. Bury St. Edmonds: Thames Valley Test Company; 1993.
Snowden JS, Neary D, Mann DMA. Fronto-temporal lobar degeneration: fronto-temporal dementia, progressive aphasia, semantic dementia. New York: Churchill Livingstone; 1996.
Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B, et al. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments.
Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group of tauopathies. [Review].
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia.
Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau.
Sumi SM, Bird TD, Nochlin D, Raskind MA, et al. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala.
Tsuang D, Raskind MA, Leverenz J, Peskind ER, Schellenberg G, Bird TD, et al. The effect of apolipoprotein E genotype on expression of an autosomal dominant schizophreniform disorder with progressive dementia and neurofibrillary tangles.
Welsh KA, Butters N, Mohs RC, Beekly D, Edland S, Fillenbaum G, et al. The consortium to establish a registry in Alzheimer's Disease (CERAD). Part V. A normative study of the neuropsychological battery.
Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG, et al. Localization of disinhibition–dementia–parkinsonism–amyotrophy complex to 17q21–22.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}