




Abstract
Quantitative gait analysis has been used to elucidate characteristic features of neurological gait disturbances. Although a number of studies compared single patient groups with controls, there are only a few studies comparing gait parameters between patients with different neurological disorders affecting gait. In the present study, gait parameters were compared between control subjects, patients with parkinsonian gait due to idiopathic Parkinson's disease, subjects suffering from cerebellar ataxia and patients with gait disturbance due to subcortical arteriosclerotic encephalopathy. In addition to recording of baseline parameters during preferred walking velocity, subjects were required to vary velocity from very slow to very fast. Values of velocity and stride length from each subject were then used for linear regression analysis. Whereas all patient groups showed slower walking velocity and reduced step length compared with healthy controls when assessed during preferred walking, patients with ataxia and subcortical arteriosclerotic encephalopathy had, in addition, increased variability of amplitude and timing of steps. Regression analysis showed that with changing velocity, subjects with Parkinson's disease changed their stride length in the same proportion as that measured in controls. In contrast, patients with ataxia and subcortical arteriosclerotic encephalopathy had a disproportionate contribution of stride length when velocity was increased. Whereas the findings in patients with Parkinson's disease can be explained as a reduction of force gain, the observations for patients with ataxia and subcortical arteriosclerotic encephalopathy reflect an altered spatiotemporal gait strategy in order to compensate for instability. The similarity of gait disturbance in subcortical arteriosclerotic encephalopathy and cerebellar ataxia suggests common mechanisms.
Introduction
Many descriptions and clinical observations deal with gait patterns in neurological disturbances of walking (for a review, see Dietz, 1997). In the early stages of idiopathic Parkinson's disease, gait is characterized mainly by reduced speed and decreased amplitude of leg movements, accompanied by reduced arm swing (Brown and Steiger, 1996). Falls and complex gait disturbances such as freezing and start hesitation are usually confined to the later stages of Parkinson's disease (Giladi et al., 1992).
In cerebellar disease, gait is slow and wide-based, with irregular timing and amplitude of steps. Sway can be omnidirectional in lesions of the vestibulocerebellum, lateralized in hemispheral lesions and predominantly anteroposterior in atrophy of the anterior lobe (Diener and Dichgans, 1996). Apart from disequilibrium, a disturbance of trunk and limb kinematics and interlimb co-ordination is presumed to be responsible for gait disturbance (Diener et al., 1992).
In subcortical arteriosclerotic encephalopathy, gait disturbance is characterized mainly by a small-stepped and broad-based gait often associated with start hesitation and disequilibrium (Giladi et al., 1997; van Zagten et al., 1998). Furthermore, disturbances of associated upper extremity and trunk movements have been described (Thompson and Marsden, 1987). The term lower body parkinsonism (Fitzgerald and Jankovic, 1989) was coined since walking appears parkinsonian, with upper limb and mimic function being relatively preserved. Clinically and pathophysiologically, gait disturbance in subcortical arteriosclerotic encephalopathy is closely related to that found in hydrocephalus and to so-called senile gait (Thompson and Marsden, 1987).
Whereas the current classifications of gait disturbances (Nutt et al., 1993) rely on descriptive clinical criteria and gestalt features, e.g. `wooden appearance' of frontal gait (Thompson and Marsden, 1987), several studies have used precision apparatus for gait analysis in order to characterize walking disorders by means of quantitative measures. However, although this approach has been used frequently in Parkinson's disease, there are only a few studies reporting quantitative measures of gait kinetics in subcortical arteriosclerotic encephalopathy and cerebellar ataxia. In Parkinson's disease, a higher step-to-step variability of stride length without disturbance of rhythmic timing of stepping has been reported (Blin et al., 1990; Vieregge et al., 1997) which does not appear to respond to l-dopa (Blin et al., 1991). Reduced velocity of walking and decreased amplitude of leg movements corresponding to clinical observations have been reported in quantitative studies on parkinsonian gait (e.g. Blin et al., 1990; Bowes et al., 1992; Morris et al., 1994). However, this finding is not specific for Parkinson's disease since reductions of stride and speed have also been described in ataxia and subcortical arteriosclerotic encephalopathy. No difference between subcortical arteriosclerotic encephalopathy and Parkinson's disease was found in a quantitative comparison of these parameters (Zijlmans et al., 1996). Taking into account that gait velocity, step length and cadence are related parameters, Morris et al. (Morris et al., 1994, 1996) assessed gait in controls and patients with Parkinson's disease while they were walking with different externally imposed velocities and cadences. A reduced stride length for any given velocity was reported in patients. In contrast to Morris et al. (Morris et al., 1998) who attributed this finding to a disorder of motor set, Elble et al. (Elbe et al., 1992) considered stride-dependent reduction of velocity as a non-specific measure without significant value in the differential diagnosis of gait disorders.
Given the latter limitations inherent in the interpretation of gait parameters and the paucity of gait studies in subcortical arteriosclerotic encephalopathy and ataxia, it remains debatable whether quantitative gait analysis yields specific features for distinct pathological conditions. The present study was thus designed to compare gait parameters at the preferred walking velocity and the relationship between step length and gait speed during changes of velocity of walking in patients with Parkinson's disease, subcortical arteriosclerotic encephalopathy and ataxia.
Material and methods
Subjects
A total of 100 subjects, comprising 30 patients with gait disturbance due to idiopathic Parkinson's disease, 20 patients with gait ataxia, 20 patients with gait disturbance due to subcortical arteriosclerotic encephalopathy and 30 age-, sex- and height-matched control subjects with no history of neurological disease, participated in the experiment (Table 1). Patients with Parkinson's disease were diagnosed according to UK brain bank criteria (Hughes et al., 1992) and showed slowing of gait reflected by a minimum score of 1 for item 29 (gait) in part III of the Unified Parkinson's Disease Rating Scale. Mean disease duration was 5.2 years (SD 4.9). Twelve patients were assessed before initiation of dopaminergic therapy (de novo Parkinson's disease) and 18 patients received regular l-dopa medication (plus adjunct therapy) and had persisting slowing of gait despite good to excellent response concerning tremor, rigidity and hypokinesia of the arms and legs. The mean daily dosage of l-dopa was 517 mg (SD 313). Five patients additionally were receiving selegiline, four bromocriptine or pergolide, three tolcapone and three amantadine.
Patients with cerebellar ataxia were referred regularly to the gait and balance laboratory for assessment of gait instability. In eight, there was cerebellar involvement due to multiple sclerosis, seven patients were diagnosed as having spinocerebellar atrophy, three had focal vascular cerebellar midline lesions and two were suffering from toxic cerebellopathy due to chronic alcoholism. All patients showed the following clinical signs: subjective disequilibrium, inability to perform tandem gait and increased omnidirectional sway during stance with eyes open and/or closed.
Subjects with gait disturbance due to subcortical arteriosclerotic encephalopathy fulfilled the clinical criteria for cautious gait or frontal gait (Nutt et al., 1993) and showed multiple subcortical white matter lesions or leukaraiosis in CT or MRI scans. Several patients had a history of transient ischaemic attack or minor stroke, but none had suffered from territorial infarctions. Five patients with subcortical arteriosclerotic encephalopathy showed signs of mild dementia that did not preclude co-operative participation. Patients with subcortical arteriosclerotic encephalopathy showing spontaneous or gaze-evoked nystagmus suggesting vestibular or cerebellar midline lesions were not included.
Patients in all groups were required to be able to walk the required distance 10 times without fatigue and to vary their gait velocity for at least 30 m/min between the slowest and fastest gait. Subjects with gait or balance problems due to orthopaedic, cardiovascular or visual disorders were not included in the study. Furthermore, patients with spastic increase of muscle tone or bilateral sensory loss in the lower extremities exceeding the reduction of distal vibration sense were not included. The study was approved by the Ethics committee of the University of Innsbruck, and informed consent according to the declaration of Helsinki was obtained from all subjects.
Apparatus
Gait parameters were recorded automatically using a device designed by Bessou et al. (Bessou et al., 1988) which has been used previously for gait analysis in idiopathic Parkinson's disease by other authors (Blin et al., 1990; Ferrandez and Blin, 1991). Longitudinal locomotor displacements are transmitted to the recording apparatus by means of threads attached by Velcro straps to each foot at the level of the head of the second metatarsal. The traction of the threads during gait leads to rotation of a pulley system monitored by an optical recording device. Signal output is transmitted to a desktop computer with appropriate software allowing calculation of stride length, duration of swing and stance time of each foot, and all measures related to these parameters (i.e. double support time, stride duration, velocity, cadence). Furthermore step-to-step variability of all parameters can be assessed. The length of the threads allows for a walking distance of 10 m without the gait being restrained by relevant resistance since the energy absorption exerted by the backward traction of the pulley only corresponds to 3% of the kinetic energy of the mass centre of an inferior limb (Bessou et al., 1988).
Procedure
Recordings were performed on a grey-coloured walkway in a quiet environment. The first two passes were not used for investigational purposes but served to familiarize subjects with the recording conditions. The subjects were then instructed to walk with different gait velocities in the following order: normal, slow, fast, normal, very slow, very fast. Additionally, static posturography was performed in patients with ataxia and in healthy controls in order to quantify and characterize the sway pattern in the former group. Posturography was performed on a force platform (Dynatronic Instruments, France). Measurements of displacement of the centre of foot pressure were taken for 51 s with the feet placed at an angle of 30°, heels 2 cm apart and eyes open. Total sway path and directional sway (lateral and anteroposterior displacements) were recorded. In order to determine the directional sway preponderance, a direction index [lateral sway path/(a – p sway); where a = anterior and p = posterior] was calculated. In three subjects with ataxia, static posturography was not possible since these patients were unable to sustain a quiet stance in the required position for the time needed for measurement.
Data analysis
The first three and the last two steps of each pass were eliminated from the data sets to eliminate the influence of acceleration and deceleration. Following the suggestions of Gabell and Nayak (Gabell and Nayak, 1984), the variability index (CoVar = SD/mean × 100) was used to express the intra-individual step-to-step variability of step length and stride duration. Since variances were not homogeneous, a non-parametric procedure (Dunn's test) was used to compare these parameters between groups using the values recorded for the second pass of preferred walking velocity. Since these measures were assumed to be dependent on velocity, no comparisons of stride length, stride duration and double support time during preferred walking velocity were performed. To establish the relationship between velocity and stride length, a linear regression analysis of these parameters was performed using the six measurements for each subject. Values for the slope and intercept were then calculated for each individual scatterplot. An example of a scatterplot for a control subject is depicted in Fig. 1. With the results of tests for equality of variance (Levene) and normal distribution (Kolmogorov–Smirnoff) being satisfactory, one-way ANOVA and Bonferroni corrected t tests for post hoc analysis were used for between-groups comparisons of slope, while intercepts (non-homogeneous variances) were compared using Dunn's test. Posturographic measures were compared using a non-parametric procedure (Mann–Whitney U test)
Results
Gait parameters during preferred walking velocity (Table 2)
Step-to-step variability was different between groups for both stride duration and step length, with patients with ataxia and subcortical arteriosclerotic encephalopathy showing increased values compared with both controls and patients with Parkinson's disease, indicating irregular timing and amplitude of stepping (P < 0.05). In patients with Parkinson's disease, timing of steps was not irregular, with values for CoVar of stride duration corresponding to control measures.
Posturography
The mean sway path was 335.1 mm (SD 74.8) in healthy controls and 830.3 mm (SD 351.9) in patients with ataxia (P < 0.001). In patients with ataxia, the direction index ranged from 0.41 to 1.24 (mean 0.785) and was thus comparable with the corresponding range established in healthy controls (0.47–1.41, mean 0.928), suggesting that sway was omnidirectional in all patients.
Regression analysis
In all subjects, the relationships of gait velocity versus stride length could be accommodated well within linear regression, with R2 > 0.8 in all and R2 > 0.9 in 95 of 100 subjects. Slopes were different between groups (P < 0.001, F 25,7) and significantly flatter in patients with ataxia and subcortical arteriosclerotic encephalopathy compared with both controls and patients with Parkinson's disease (P < 0.05). No differences were observed between patients with subcortical arteriosclerotic encephalopathy and ataxia and between patients with Parkinson's disease and controls. Figure 2 shows the distribution of slope values for each group. Intercept values also differed between groups, with controls having lower intercepts than all patient groups, and patients with Parkinson's disease showing lower intercepts than patients with subcortical arteriosclerotic encephalopathy and ataxia (P < 0.05). Figure 3 shows the distribution of the individual intercept values for all four groups. It can be deduced that patients with Parkinson's disease (showing slopes corresponding to control values) have a proportionally reduced amplitude of stride length at any given velocity compared with controls. A direct comparison of intercepts between patients with Parkinson's disease or controls and patients with subcortical arteriosclerotic encephalopathy or ataxia is precluded since intercept values are not independent of slope and thus are increased disproportionately in the latter groups. The regression lines corresponding to the mean slope and intercept values for all subjects are shown in Fig. 4. All patient groups showed reduced stride length compared with controls in the velocity range corresponding to the preferred walking pattern, but with different relative changes of stride length when gait velocity was modified. Patients with multiple sclerosis were found to have slightly lower values for slope (72.1, SD 10) and higher intercepts (–25.2 m/min, SD 15) compared with the other patients with ataxia (slope 83.3, SD 15.9; intercept –34, 5 m/min, SD 19.2), but no qualitative differences emerged.
Discussion
In contrast to Parkinson's disease, quantitative studies of gait kinetics have been performed only rarely in ataxia and subcortical arteriosclerotic encephalopathy (Hennerici et al., 1994; Zijlmans et al., 1996; Palliyath et al., 1998). In the present study, comparison of gait parameters between these patient groups showed reduced amplitude of stride during preferred walking velocity in all patients compared with controls. In contrast to patients with Parkinson's disease, the relative reduction of stride length compared with controls was not constant, but changed with gait velocity in patients with ataxia and subcortical arteriosclerotic encephalopathy. Furthermore, patients with ataxia and subcortical arteriosclerotic encephalopathy showed increased step-to-step variability of stride duration and step length. Patients with ataxia showed irregular amplitudes and movement times which were consistent with clinical observations and the failures of relative timing and force generation in multijoint movements, presumed to occur in cerebellar dysfunction (Hallet and Massaquoi, 1993; Palliyath et al., 1998). In subcortical arteriosclerotic encephalopathy, irregular timing and amplitudes of stride have not been reported so far. Neither of the quantitative gait studies previously performed in patients with subcortical arteriosclerotic encephalopathy assessed step-to-step variabilities of gait parameters, but were limited to the recording of mean values for stride length and duration during preferred walking velocity (Hennerici et al., 1994; Zijlmans, 1996). In patients with Parkinson's disease, some increase of step length variability without disturbance of rhythmic timing, as reported by other authors (Blin et al., 1990; Vieregge et al., 1997), was confirmed, although statistical significance was not reached in a multiple group comparison. The present results suggest that irregular step length is not a distinct feature in Parkinson's disease, but can be found even more pronounced in ataxia and gait disturbance due to subcortical arteriosclerotic encephalopathy.
Since the finding of reduced step length during preferred walking velocity was anticipated and assumed to be non-specific, regression analysis of velocity versus step length was used to analyse the spatiotemporal characteristics of gait. Various previous studies performing regression analysis in healthy subjects have shown that the relationship between speed and step length is linear, with small differences between the sexes and different age groups (Grieve, 1968; Hirokawa, 1989). The linear relationship of velocity to stride length is only consistent for the medium velocity range (Larsson et al., 1980), with a flattening (Bowes et al., 1992) or even a break point (Hirokawa, 1989) occurring when the individual upper speed limits are approached.
In the present study, the linear relationship between stride length and walking velocity, as reflected by high R2 values, was demonstrated for all subjects and disclosed distinct abnormalities of gait motor control in patients with Parkinson's disease compared with the other patient groups. In patients with Parkinson's disease, slopes were comparable with control measures, whereas higher intercepts indicated a reduced stride amplitude at any given velocity. This result is in keeping with the findings of Morris et al. (Morris et al., 1994) and Ferrandez and Blin (Ferrandez and Blin, 1991). In a recent study by Morris et al. (Morris et al., 1998), subjects were instructed to adapt their cadence to a metronome signal set at rates ranging from 40 to 180 steps/min. Comparison of linear regression of stride length and velocity between healthy controls and 20 patients with Parkinson's disease showed that slopes did not differ significantly between groups, whereas higher mean intercepts were found in patients. Administration of l-dopa led to lower intercepts, with the slope remaining unchanged. The results of the present study show that this behaviour is not confined to metronome walking but also occurs with freely chosen speed and cadence.
In contrast to those with Parkinson's disease, patients with ataxia showed a disproportionate decrease in step length occurring during slow walking and a disproportionate increase occurring during fast walking. Cerebellar pathology was heterogeneous in the patient group suffering from ataxia, and included suspected anterior lobe lesions in cerebellopathy due to alcoholism, vermal lesions due to ischaemia and both cerebellar hemispheral and midline lesions due to multiple sclerosis. Although ataxia was the major complaint in the latter group and any patients with clinically significant sensory disturbance and/or spasticity affecting the ability to walk were excluded, additional extracerebellar lesions were demonstrated by MRI in all patients and may have contributed to the gait instability. In spite of this and although affected cerebellar structures in the ataxia group subserve different neural circuits (Diener and Dichgans, 1996), all patients showed a rather uniform clinical picture of disequilibrium with gait disturbance and omnidirectional postural instability quantified by means of gait analysis and static posturography. With the slope of velocity versus stride being altered regardless of the aetiology of ataxia, the disproportionate contribution of step length to the increase of velocity in these patients is likely to reflect a non-specific feature of disequilibrium rather than dysfunction of a defined neural structure.
The assumption of a non-specific mechanism being responsible for the observed gait pattern is supported further by the finding that regression equations in patients with subcortical arteriosclerotic encephalopathy were similar to those in cerebellar disease. The mechanisms by which diffuse white matter disease interferes with gait are not clearly understood. Lesioning of periventricular fibres connecting the basal ganglia and supplementary motor area are believed to play a significant role (Thompson and Marsden, 1987; Curran and Lang, 1994). The neurophysiological substrate for ataxic features of the gait disorder due to subcortical arteriosclerotic encephalopathy is probably heterogeneous, including lesion of the periventricular fibres connecting the cerebellum and motor cortex (Thompson and Marsden, 1987), impaired central somatosensory integration (Baloh et al., 1995) and disruption of long-loop reflexes (Masdeu et al., 1989). Atrophy of midline cerebellar structures is sometimes found in subcortical arteriosclerotic encephalopathy but not consistently related to disequilibrium (Masdeu et al., 1989; Baloh et al., 1995). The heterogeneity of neural damage in subcortical arteriosclerotic encephalopathy again strongly argues against a distinct neural substrate being causative for the gait pattern observed in the present study.
A possible explanation for the changes of slope for velocity versus stride described in the present study is a compensatory response to omnidirectional instability, analogous to the broadening of base. During slow gait, equilibrium is challenged by lateral sway vectors forcing the affected subject to reduce periods of single leg support disproportionally in order to keep lateral instability minimal. With increasing velocity, the sagittal vector becomes predominant and large steps are needed to prevent the subject from falling forward. This hypothesis is in line with previous suggestions, made on theoretical grounds, that deficits of neuromuscular coordination or postural instability might lead to compensatory alteration of the relationship between velocity and stride length. (Grieve, 1968; Larsson et al., 1980).
In contrast to the clinical descriptions of gait disturbance in subcortical arteriosclerotic encephalopathy stressing the similarity to parkinsonian gait, as reflected by the terms `lower body parkinsonism' or `vascular parkinsonism', the present findings emphasize the relationship to ataxia. Clinically ataxic signs of gait in subcortical arteriosclerotic encephalopathy, including broad-based walking and instability with increased likelihood of falling, have been described by several authors (Thompson and Marsden, 1987; Masdeu et al., 1989; Baloh et al., 1995). The results of the present study suggest that the similarity between ataxia and subcortical arteriosclerotic encephalopathy is not limited to these clinical features but also includes irregular gait patterning and altered regulation of gait velocity. The finding that step length contributed disproportionately to the increase of gait velocity in the present study is complementary to the observation of Thompson and Marsden (Thompson and Marsden, 1987) that festination (i.e. rapid short stepping) is not found in patients with subcortical arteriosclerotic encephalopathy.
Conclusion
The present study shows that the clinical sign of short-stepped gait at preferred walking velocity can be due to two different pathological patterns of gait regulation. The first, found in patients with Parkinson's disease, was characterized by a proportionally decreased stride length for any given gait velocity, with mild irregularity of stride amplitude and no signs of arrhythmicity. This pattern corresponds to decreased force gain and allows minimization of energy expenditure The second pattern, observed in patients with ataxia and subcortical arteriosclerotic encephalopathy, was characterized by a disproportionately reduced stride length with slow gait and a disproportionate increase of stride length with higher walking speed. This pattern provides evidence for dynamic equilibrium in the presence of increased omnidirectional sway. Irregular scaling and timing of stride were associated with the latter pattern. The present approach, i.e. linear regression analysis of gait parameters, was shown to provide enhanced information about gait motor control and should be included in studies assessing other disturbances of walking or protocols dealing with rehabilitation assessments, drug monitoring or follow up observations of gait disturbances.
Subject characteristics for gait analysis
| Group | n | Male/female | Age (years) | Height (cm) |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Controls | 30 | 19/11 | 60.9 (8.0) | 171.8 (9.1) |
| Parkinson | 30 | 19/11 | 65.0 (9.3) | 168.9 (7.5) |
| Ataxia | 20 | 13/7 | 41.4 (14.2) | 169.2 (8.1) |
| SAE | 20 | 11/9 | 73.7 (11.8) | 165.1 (5.0) |
| Group | n | Male/female | Age (years) | Height (cm) |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Controls | 30 | 19/11 | 60.9 (8.0) | 171.8 (9.1) |
| Parkinson | 30 | 19/11 | 65.0 (9.3) | 168.9 (7.5) |
| Ataxia | 20 | 13/7 | 41.4 (14.2) | 169.2 (8.1) |
| SAE | 20 | 11/9 | 73.7 (11.8) | 165.1 (5.0) |
Subject characteristics for gait analysis
| Group | n | Male/female | Age (years) | Height (cm) |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Controls | 30 | 19/11 | 60.9 (8.0) | 171.8 (9.1) |
| Parkinson | 30 | 19/11 | 65.0 (9.3) | 168.9 (7.5) |
| Ataxia | 20 | 13/7 | 41.4 (14.2) | 169.2 (8.1) |
| SAE | 20 | 11/9 | 73.7 (11.8) | 165.1 (5.0) |
| Group | n | Male/female | Age (years) | Height (cm) |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Controls | 30 | 19/11 | 60.9 (8.0) | 171.8 (9.1) |
| Parkinson | 30 | 19/11 | 65.0 (9.3) | 168.9 (7.5) |
| Ataxia | 20 | 13/7 | 41.4 (14.2) | 169.2 (8.1) |
| SAE | 20 | 11/9 | 73.7 (11.8) | 165.1 (5.0) |
Gait parameters at preferred walking velocity
| Group | Velocity (m/min) | Cadence (steps/min) | CoVar step length | CoVar stride duration |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Control | 61.9 (6.0) | 98.5 (7.3) | 2.8 (1.2) | 2.3 (1.2) |
| Parkinson | 49.2 (9.5) | 94.5 (7.7) | 3.7 (1.9) | 2.4 (0.9) |
| Ataxia | 44.8 (11.7) | 93.1 (11.1) | 7.7 (5.5) | 4.8 (2.1) |
| SAE | 32.5 (9.2) | 94.7 (11.1) | 9.2 (4.9) | 3.9 (1.7) |
| Group | Velocity (m/min) | Cadence (steps/min) | CoVar step length | CoVar stride duration |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Control | 61.9 (6.0) | 98.5 (7.3) | 2.8 (1.2) | 2.3 (1.2) |
| Parkinson | 49.2 (9.5) | 94.5 (7.7) | 3.7 (1.9) | 2.4 (0.9) |
| Ataxia | 44.8 (11.7) | 93.1 (11.1) | 7.7 (5.5) | 4.8 (2.1) |
| SAE | 32.5 (9.2) | 94.7 (11.1) | 9.2 (4.9) | 3.9 (1.7) |
Gait parameters at preferred walking velocity
| Group | Velocity (m/min) | Cadence (steps/min) | CoVar step length | CoVar stride duration |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Control | 61.9 (6.0) | 98.5 (7.3) | 2.8 (1.2) | 2.3 (1.2) |
| Parkinson | 49.2 (9.5) | 94.5 (7.7) | 3.7 (1.9) | 2.4 (0.9) |
| Ataxia | 44.8 (11.7) | 93.1 (11.1) | 7.7 (5.5) | 4.8 (2.1) |
| SAE | 32.5 (9.2) | 94.7 (11.1) | 9.2 (4.9) | 3.9 (1.7) |
| Group | Velocity (m/min) | Cadence (steps/min) | CoVar step length | CoVar stride duration |
|---|---|---|---|---|
| Values are mean (SD). SAE = subcortical arteriosclerotic encephalopathy. | ||||
| Control | 61.9 (6.0) | 98.5 (7.3) | 2.8 (1.2) | 2.3 (1.2) |
| Parkinson | 49.2 (9.5) | 94.5 (7.7) | 3.7 (1.9) | 2.4 (0.9) |
| Ataxia | 44.8 (11.7) | 93.1 (11.1) | 7.7 (5.5) | 4.8 (2.1) |
| SAE | 32.5 (9.2) | 94.7 (11.1) | 9.2 (4.9) | 3.9 (1.7) |
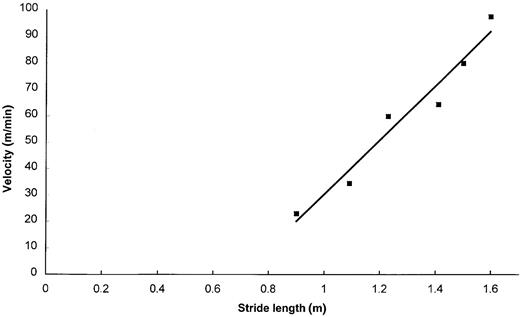
Diagram showing the linear regression of velocity versus stride in a control subject (y = 102.35x – 71.932; R2 = 0.9561).
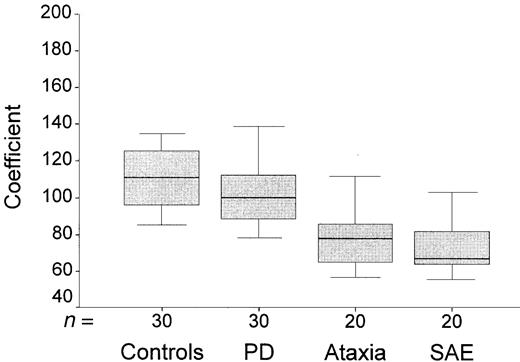
Box-plots showing the distribution of slopes for velocity versus stride in controls and patient groups. Medians, ranges, and 25 and 75 percentiles are shown. PD = Parkinson's disease; SAE = subcortical arteriosclerotic encephalopathy
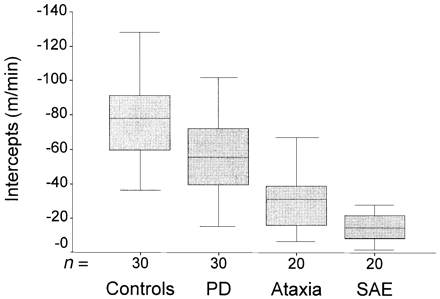
Box-plots showing the distribution of intercepts for velocity versus stride in controls and patient groups. Medians, ranges, and 25 and 75 percentiles are shown. PD = Parkinson's disease; SAE = subcortical arteriosclerotic encephalopathy
![Diagram showing the mean regressions of velocity versus stride in controls (y = 113x – 78) and patient groups [subcortical arteriosclerotic encephalopathy (SAE), y = 73x –16; ataxia, y = 79x –31; Parkinson's disease (PD), y = 112x – 64].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/122/7/10.1093_brain_122.7.1349/1/m_b071004.gif?Expires=1716327368&Signature=qlMPOVrCqtxzG9PgDkpgUHkpQHdU~bMb6ubZD88uYM1U4dPQily9sionT9eoFbo7nKMIn2MZuutA0gEbxW1QU08g4TdsYQiWWI7rT2Mv59hng0DvD~q8HQnAGJVlise0pwjj~Bgxao9U9eNLysFk4Urto8NKeZ7RwKrWOXTVZkV~Ktui0hSONBtwueIYv34xkFtZSU-cvbJRLwdfQY85lHxGYKq6AnR-CsbQn9YHLAjN6tVO7G4s9op0C~mxn47CUV7unS2B~lQQclUhxyZmdATDN-7bD94eC3TaKl6P1dDKsUsQsltQ2jbV5lNVF~JCw10si~wIRtzdZ92srUTjkA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Diagram showing the mean regressions of velocity versus stride in controls (y = 113x – 78) and patient groups [subcortical arteriosclerotic encephalopathy (SAE), y = 73x –16; ataxia, y = 79x –31; Parkinson's disease (PD), y = 112x – 64].
References
Baloh RW, Yue Q, Socotch TM, Jacobson KM. White matter lesions and disequilibrium in older people.
Bessou P, Dupui P, Montoya R, Pages B. Simultaneous recording of longitudinal displacements of both feet during human walking. J Physiol (Paris) 1988–89; 83: 102–10.
Blin O, Ferrandez AM, Serratrice G. Quantitative analysis of gait in Parkinson patients: increased variability of stride length.
Blin O, Ferrandez AM, Pailhous J, Serratrice G. Dopa-sensitive and dopa-resistant gait parameters in Parkinson's disease.
Bowes SG, Charlett A, Dobbs RJ, Lubel DD, Mehta R, O'Neill CJ, et al. Gait in relation to ageing and idiopathic parkinsonism.
Brown P, Steiger MJ. Basal ganglia gait disorders. In: Bronstein AM, Brandt T, Woollacott MH, editors. Clinical disorders of balance, posture and gait. London: Arnold; 1996. p. 156–67.
Curran T, Lang AE. Parkinsonian syndromes associated with hydrocephalus: case reports, a review of the literature, and pathophysiological hypotheses. [Review].
Diener HC, Dichgans J. Cerebellar and spinocerebellar gait disorders. In: Bronstein AM, Brandt T, Woollacott MH, editors. Clinical disorders of balance, posture and gait. London: Arnold; 1996. p. 147–55.
Diener HC, Dichgans J, Guschlbauer B, Bacher M, Rapp H, Klockgether T. The coordination of posture and voluntary movement in patients with cerebellar dysfunction.
Dietz V. Neurophysiology of gait disorders: present and future applications. [Review].
Ferrandez AM, Blin O. A comparison between the effect of intentional modulations and the action of l-dopa on gait in Parkinson's disease.
Fitzgerald PM, Jankovic J. Lower body parkinsonism: evidence for vascular etiology.
Giladi N, McMahon D, Przedborski S, Flaster E, Guillory S, Kostic V, et al. Motor blocks in Parkinson's disease.
Giladi N, Kao R, Fahn S. Freezing phenomenon in patients with parkinsonian syndromes.
Hallett M, Massaquoi SG. Physiologic studies of dysmetria in patients with cerebellar deficits. [Review].
Hennerici MG, Oster M, Cohen S, Schwartz A, Motsch, Daffertshofer M. Are gait disturbances and white matter degeneration early indicators of vascular dementia?
Hirokawa S. Normal gait characteristics under temporal and distance constraints.
Hughes AJ, Ben-Shlomo Y, Daniels SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson's disease: a clinicopathologic study [published erratum appears in Neurology 1992; 42: 1436] [see comments].
Larsson LE, Odenrick P, Sandlund B, Weitz P, Öberg PA. The phases of the stride and their interaction in human gait.
Masdeu JC, Wolfson L, Lantos G, Tobin JN, Grober E, Whipple R, et al. Brain white-matter changes in the elderly prone to falling [see comments].
Morris ME, Iansek R, Matyas TA, Summers JJ. Ability to modulate walking cadence remains intact in Parkinson's disease.
Morris ME, Iansek R, Matyas TA, Summers JJ. Stride length regulation in Parkinson's disease. Normalization strategies and underlying mechanisms.
Morris M, Iansek R, Matyas T, Summers J. Abnormalities in stride length–cadence relation in parkinsonian gait.
Nutt JG, Marsden CD, Thompson PD. Human walking and higher-level gait disorders, particularly in the elderly [see comments].
Palliyath S, Hallett M, Thomas SL, Lebiedowska MK. Gait in patients with cerebellar ataxia.
Thompson PD, Marsden CD. Gait disorder of subcortical arteriosclerotic encephalopathy: Binswanger's disease.
van Zagten M, Lodder J, Kessels F. Gait disorder and parkinsonian signs in patients with stroke related to small deep infarcts and white matter lesions.
Vieregge P, Stolze H, Klein C, Heberlein I. Gait quantitation in Parkinson's disease—locomotor disability and correlation to clinical rating scales.


{kind=link}
{kind=link}
{kind=link}
{kind=link}