




Abstract
Leber's hereditary optic neuropathy (LHON) is a common cause of bilateral optic nerve disease. The majority of LHON patients harbour one of three point mutations of the mitochondrial DNA (mtDNA) complex I, or NADH:ubiquinone oxidoreductase (ND) genes (G11778A in ND4, G3460A in ND1, T14484C in ND6). As a consequence, screening for these mutations has become part of the routine clinical investigation of young adults who present with bilateral optic neuropathy, and the absence of these mutations is interpreted as indicating there is a low likelihood that an optic neuropathy is LHON. However, there are many individuals who develop the clinical features of LHON but who do not harbour one of these primary LHON mutations. We describe two LHON pedigrees that harbour the same novel point mutation within the mtDNA ND6 gene (A14495G). This mutation was heteroplasmic in both families, and sequencing of the mitochondrial genome confirmed that the mutation arose on two independent occasions. This is the seventh mutation in the ND6 gene that causes optic neuropathy, indicating that this gene is a hot spot for LHON mutations. Protein modelling studies indicate that all of these pathogenic mutations lie within close proximity to one another in a hydrophobic cleft or pocket. This is the first evidence for a relationship between a specific disease phenotype and a specific structural domain within a mitochondrial respiratory chain subunit. These findings suggest that the mtDNA ND6 gene should be sequenced in all patients with LHON who do not harbour one of the three common LHON mutations.
Introduction
Leber's hereditary optic neuropathy (LHON, MIM, Mendelian Inheritance in Man 535000) is a common cause of subacute bilateral optic neuropathy that usually presents in early adult life and that preferentially affects males (Leber, 1871; Riordan-Eva et al., 1995). In the vast majority of cases, LHON is due to one of three mitochondrial DNA (mtDNA) point mutations in the NADH:ubiquinone oxidoreductase (ND) genes that encode complex I subunits of the mitochondrial respiratory chain (Mackey et al., 1996). The G11778A mutation results in an arginine→histidine substitution at amino acid position 340 (R340H) of the ND4 subunit (Wallace et al., 1988), the G3460A mutation causes an alanine→threonine (A52T) substitution in the ND1 subunit (Howell et al., 1991), and the T14484C mutation causes a methionine→valine (M64V) substitution in the ND6 subunit (Johns et al., 1992).
The investigation of patients with suspected LHON involves allele-specific screening for one of the three primary LHON mutations. The identification of a primary LHON mutation has profound implications for the family, and there are well-established recurrence risks for matrilineal lineages that carry the G11778A or T14484C LHON mutations (Harding et al., 1995; Macmillan et al., 1998). There are substantial numbers of patients with LHON who do not carry one of these primary mutations (Mackey et al., 1996). This situation generates considerable uncertainty in the clinic, particularly if it is not possible to identify some of the characteristic phenotypic features of LHON during the acute phase, or if there is only a small number of affected individuals within the family. Genetic counselling is especially difficult for apparent singleton cases, in which the neuropathy may be clinically indistinguishable from sporadic or autosomally inherited optic atrophy.
Here we report two families that presented with the clinical features of LHON, but who did not harbour the G11778A, G3460A or T14484C mutation. Sequence analysis of their mtDNA revealed a pathogenic mutation in the mtDNA ND6 gene within a highly conserved hydrophobic cleft that appears to be a hot spot for LHON mutations. These studies indicate that the ND6 gene should be sequenced in patients who may have LHON but who lack a known pathogenic mutation.
Patients and methods
Clinical features
British family (for pedigree, see Fig. 1)
Family member II-2.
A 53-year-old woman, who did not smoke and took <4 U of alcohol per week, developed painless blurring of vision in the right eye at 30 years of age, followed 3 months later by painless visual loss in her left eye over a few days. Routine haematological and biochemical investigations at the time revealed a macrocytic anaemia (further details are not available). A brain CT scan was normal, as was a lumbar puncture. CSF electrophoresis did not identify any oligoclonal immunoglobulin bands that were not matched in the serum. She was treated empirically with parenteral vitamin B12 but did not report any improvement. On recent assessment, her visual acuity was 1/60 bilaterally with bilateral central scotomata on visual field testing, and fundoscopy showed bilateral optic atrophy.
Family member II-3.
An asymptomatic 53-year-old woman.
Family member III-1.
This 28-year-old man developed bilateral, painless sequential visual loss at 10 years of age with associated optic disc swelling. He was initially educated at a school for the visually impaired, but throughout his teenage years his vision improved and he returned to mainstream schooling. On recent assessment, his visual acuity had improved to 6/9 in each eye and he could read all of the Ishihara test plates. Fundoscopy revealed bilateral temporal disc pallor.
Family member III-2.
In August 1998, at the age of 26 years, this individual developed painless central visual loss affecting his left eye over a period of 2 months. In January 1999, similar visual loss developed in his right eye. In mid-February, visual acuity was 6/9 in the right eye, with a small central scotoma, and counting fingers in the left eye, with a larger central scotoma. With the right eye, he could only identify the test plate of the Ishihara series. Around the right optic disc there was swelling of the nerve fibre layer and a few telangiectatic superficial retinal vessels (Fig. 2A and B). There was no disc leakage on fluorescein angiography. There was temporal pallor of the left optic disc with corresponding atrophy of the nerve fibre layer. The patient smoked 10–20 cigarettes per day and consumed moderate amounts of alcohol. He admitted to a very poor diet. Serum vitamin B12, red cell folate and bone marrow examination were normal. Empirical treatment was started with parenteral vitamin B12, oral folate and B vitamins (including thiamine), followed by high-dose systemic steroid therapy. By the end of March 1999, vision was reduced to counting fingers on both sides with large central scotomas. Cranial MRI revealed a non-enhancing high signal in the left optic nerve (Fig. 2C), without any white matter lesions. The CSF was acellular with a normal protein and glucose content. There were no unmatched oligoclonal bands in the CSF. Visual electrophysiology confirmed bilateral optic nerve disease.
Family member III-3.
An asymptomatic 35-year-old man.
Family member III-4.
An asymptomatic 33-year-old woman.
Canadian family
A 52-year-old man presented at 17 years of age with subacute bilateral visual failure. After an initial nadir, his acuity has slowly but steadily improved over a 20-year period, although he has remained registered blind to the present time. On recent assessment at age 51 years, his acuity was limited to counting fingers. He had sluggish pupillary responses, but no relative afferent pupillary defect. Visual field testing revealed large centrocaecal scotomas, and fundoscopy confirmed bilateral optic atrophy. Recent assessment at the age of 52 years revealed no significant change in acuity or visual fields compared with previous assessments. A maternal uncle was similarly affected, but no clinical data were available.
Each subject gave informed consent to be part of the study.
Molecular genetic studies
mtDNA sequencing
Total genomic DNA was extracted from leucocytes by standard techniques.
Newcastle.
The entire mitochondrial genome for one member of the British LHON family (III-2) was amplified by PCR (polymerase chain reaction) using 25 overlapping –21 M13 forward and Reverse M13 reverse-tagged primer pairs (Andrews et al., 1999). PCR products were purified (QIAquick, Qiagen Hilden, Germany), and both strands were sequenced using a Big-Dye terminator sequencing kit (Perkin-Elmer) and an ABI 377 automated DNA sequencer.
Galveston.
DNA from the Canadian patient was analysed in Galveston. Small PCR-amplified mtDNA fragments of ~300 base pairs amplified from the Canadian patient's DNA were cloned into M13 sequencing vectors and the mtDNA sequence was determined using a manual dideoxy chain terminating method as described previously (Howell et al., 1995) (for details of the regions sequenced, see Appendix I).
Sequences were compared with the revised Cambridge Reference Sequence (rCRS) (Anderson et al., 1981; Andrews et al., 1999).
Assessment of heteroplasmy
Newcastle.
The mutation at nucleotide 14 495 did not lead to either a gain or loss of a restriction endonuclease site. We therefore designed a fluorescent primer extension assay to determine the proportion of mutant and wild-type mtDNA using a modified method of Ghosh and colleagues (Ghosh et al., 1996) and Fahy and colleagues (Fahy et al., 1997). In brief, a fluorescence-labelled primer was designed to bind specifically to the DNA sequence immediately adjacent to position 14 495. If the fluorescent primer is added to a PCR product incorporating nucleotide 14 495, along with a specific chain-extending deoxynucleotide and a specific chain-terminating dideoxynucleotide, a DNA polymerase will extend the primer by one base when it binds to the wild-type template, and by two bases when it binds to the mutant template. The primer extension assay products are then separated by electrophoresis and detected using a fluorescent DNA sequencer, and the size and relative amount of each product can be determined (for detailed methods, see Appendix II).
Galveston.
The proportion of mutated mtDNA was estimated by direct sequencing of multiple independent M13 clones.
Mutation screening strategy and polymorphic variability within the ND6 gene
The region of the ND6 gene that spans nucleotide 14 495 and the previously identified ND6 LHON mutations was sequenced in the following individuals: 112 normal control individuals, mainly of Caucasian origin; 32 individuals with neurological disease (suspected mitochondrial disease and neurodegenerative disorders including Alzheimer's disease, idiopathic Parkinson's disease and amyotrophic lateral sclerosis); 58 individuals with a clinical diagnosis of ophthalmological disease, including non-LHON optic atrophy, retinitis pigmentosa, Stargardt's disease and macular dystrophy; and 82 individuals from different pedigrees with the G11778A, G3460A or T14484C primary LHON mutations.
Analysis of ND6 secondary and tertiary structure
All of the available ND6 sequences were downloaded from the Mitbase web site (http://www3.ebi.ac.uk/Research/Mitbase/mitbase.pl) (Lanave et al., 1999) and aligned using Genedoc v2.5 (Nicholas and Nicholas, 1997) and http://www.cris.com/~ketchup/genedoc.shtml). Structural analysis was carried out using the TMpred program, accessible through the Baylor College of Medicine/Human Genome Sequencing Center web site (http://dot.imgen.bcm.tmc.edu:9331/seq-search/struc-predict.html) and the program ISREC (Swiss Institute for Experimental Cancer Research) (http://www.ch.embnet.org/), which implements the Kyte–Doolittle method for hydropathy analysis.
Results
Analysis of mtDNA sequences
The G11778A, G3460A and T14484C primary LHON mutations were not detected in the British and Canadian LHON families. There were nine differences between the mtDNA coding sequence for members of the British pedigree and the rCRS (Table 1). No other previously identified pathogenic mtDNA mutations were detected. Primer extension assay confirmed that the A14495G mutation was heteroplasmic in all of the British family members tested. The percentage of mutated mtDNA in the leucocyte/platelet DNA ranged from 23 to 93, with the highest levels in the two clinically affected males (Fig. 1). Sequencing of multiple clones from the Canadian patient confirmed that this patient was also heteroplasmic, and 10 of 12 clones (83%) carried the mutation. The Canadian mtDNA also carried an additional heteroplasmic silent polymorphism at nucleotide 11 149 [ND4/LEU130 was unchanged, five of nine clones (55%) carrying the polymorphic allele].
We also obtained the entire sequence of the 1.1-kilobase non-coding control region (D-loop) for both mtDNAs (Table 1). The Canadian LHON patient's control region sequence changes from the rCRS were T16519C and A263G. The British LHON patient's control region sequence changes from the rCRS were G16390A, T16519C, T152C and A263G. Both families clearly belonged to European haplogroup H because they carried the H haplogroup-specific allele at nucleotide 14 766 and because they lacked any other European haplogroup-specific polymorphisms (Torroni et al., 1996). However, the two mtDNA sequences differed from each other at six sites within the coding region and at two sites within the D-loop (Table 1). This confirmed that the two 14 495 LHON families had genetically distinct mtDNA backgrounds (Table 1).
The Canadian patient also included the putative secondary LHON mutation at nucleotide 4216 (Johns and Berman, 1991). It is now recognized that this is a relatively ancient European-specific polymorphism which arose in the common ancestor to haplogroups J and T (Howell et al., 1995; Torroni et al., 1996), and it is very unlikely to have any pathogenic role in LHON. The Canadian mtDNA did not carry any other characteristic polymorphisms for haplogroups J and T, and we conclude that the 4216 polymorphism is benign and that it has arisen independently in the haplogroup H background (i.e. this is a homoplastic polymorphism).
Polymorphic variability, evolutionary conservation and hydropathy plot analysis of the human ND6 gene
The A14495G mutation was not detected in the 284 normal and disease controls. In order to establish the importance of the region of the ND6 gene affected by the A14495G mutation, we also studied the sequence conservation and the predicted tertiary structure of the ND6 polypeptide between and within species. A simple hydropathy plot analysis of the ND6 subunit indicated four, or possibly five, hydrophobic domains (Fig. 3). The A14495G mutation altered one of the two most highly conserved amino acid residues of the B/C domain of ND6. Leucine-60 is invariant in vertebrates (26 mammalian species, 27 avian species and four fish species). The A14495G mutation changes both side-chain hydrophobicity and volume, replacing the large, non-polar hydrophobic leucine with the small and uncharged, but hydrophilic, serine. As a result, this substitution locally alters the Kyte–Doolittle hydropathy profile for ND6 (Fig. 3).
To establish the importance of this region in humans, we also sequenced the region of the ND6 gene around the A14495G mutation (amino acid residues 25–74, nucleotides 14 452–14 601) in 271 disease and normal controls. In this particular analysis, those patients with known pathogenic mutations in this region of the ND6 gene were excluded. Within this group, we observed 10 differences from the rCRS between nucleotides 14 452 and 14 601 (Table 2). Eight of these nucleotide changes did not affect the predicted amino acid sequence of the protein and, thus, ~80% of the polymorphisms in this region were silent. In two healthy control individuals, the nucleotide substitutions led to alterations in the predicted amino acid sequence (A14577G/I33V and A14582G/V31A). An extensive survey of the available web-based databases and the published literature revealed only one other previously described polymorphism altering the protein secondary structure within this region (T14502C/I58V) (Ozawa et al., 1991). These three substitution polymorphisms alter less conserved residues in this region of the ND6 subunit.
Discussion
A significant number of individuals who are suspected to have LHON do not harbour one of the three primary mtDNA LHON mutations. Investigation of these patients is difficult for several reasons. First, it is not possible to rely upon biochemical studies of mitochondrial dysfunction. Although the three primary LHON mutations all affect different complex I subunit genes, these mutations are not associated in a consistent pattern with a respiratory chain abnormality that can be detected in assays of isolated mitochondria or in in vivo studies (Larsson et al., 1991; Cock et al., 1995; Howell, 1997; Lodi et al., 1997; Brown, 1999). Secondly, the interpretation of mtDNA sequence data may be complex. mtDNA is highly polymorphic, and any two unrelated individuals usually harbour between 20 and 60 sequence variants (Chinnery et al., 1999). In addition, the mtDNAs of many individuals, including the Cambridge Reference Sequence (Anderson et al., 1981; Andrews et al., 1999), harbour unique or `private' polymorphisms. This genotypic variability makes it difficult to interpret novel base changes in a patient with a suspected mtDNA disease such as LHON. In other mtDNA diseases, a mixture of mutated and wild-type genomes (heteroplasmy) occurs within the same individual. It is often possible to show that high percentages of mutated mtDNA are associated with disease at the level of the family, or with a biochemical defect affecting single cells. Unfortunately, heteroplasmy is an unusual finding in families with LHON (Smith et al., 1993).
We have identified a novel mtDNA ND6 mutation at nucleotide 14 495 in two genealogically unrelated families with LHON. The mutation was heteroplasmic in both families, and there was a broad correlation between the percentage of mutated mtDNA in blood and the clinical phenotype. This base change alters a highly conserved amino acid in the C-domain (Fig. 3) of the ND6 subunit of complex I of the mitochondrial respiratory chain (Fearnley and Walker, 1992). Unlike other LHON mutations, the amino acid substitution noticeably alters the ND6 hydropathy plot, a result portending catalytic dysfunction. The mutation was not present in 112 healthy control subjects, 90 disease control subjects and 82 individuals who harboured one of the three primary LHON mutations. All of this evidence strongly indicates that the 14 495 mutation is pathogenic and that it causes LHON.
Pathogenic mechanisms of the A14495G mutation
The vast majority of pathogenic mutations associated with LHON involve the mtDNA complex I genes (Howell, 1994). Complex I is the largest enzyme complex of the mitochondrial respiratory chain. It has a boot-shaped structure, with an anchoring hydrophobic portion buried within the inner mitochondrial membrane, and the other portion, which includes the NADH dehydrogenase domain of the complex, protruding into the mitochondrial matrix (Grigorieff, 1998). The membrane arm includes all of the seven mtDNA-encoded subunits, and the matrix arm includes most of the nuclear-encoded subunits (~35 in number) and most of the prosthetic groups involved in the ND reactions (Smeitink and van den Heuvel, 1999). With the exception of the ND1 subunit, which appears to contribute to the quinone binding site (Friedrich et al., 1990), the function of the individual mitochondrially encoded complex I subunits is not known, but it is likely that they are involved in proton translocation and in the reduction of quinone (Walker, 1992).
The B/C hydrophobic domain is much longer than that required for a single, simple transmembrane helix, and our analysis of ND6 with the TMpred software (see Patients and methods) yielded a strongly preferred model in which there are six transmembrane helices oriented as shown in Fig. 4. This model resolves the B/C domain into two transmembrane helices with a short (seven amino acid residues) connecting extramembrane loop. More importantly, this model predicts that all of the pathogenic mutations are in relatively close spatial proximity, apparently defining two sides of a functionally important channel or pocket between these two helices. Our extensive sequence analysis of the B/C domain of ND6 confirms that this region is highly conserved within the Caucasian population (Table 2), presumably due to the structural and functional sensitivity of this domain to amino acid alterations.
The A14495G/L60S LHON mutation may affect proton translocation or quinone redox catalysis directly, or it may indirectly affect ND6 assembly. It is known from analysis of a frame-shift mutation in the gene that the ND6 subunit is essential for the assembly of the membrane arm of complex I (Bai and Attardi, 1998). However, we suspect that a single amino acid substitution, even the serine-for-leucine replacement, is unlikely to have such a profound effect on ND6 structure as to destabilize the complex. Recent biochemical studies have shown that another ND6 mutation (T14484C/M64V) increases the sensitivity of complex I to inhibitors binding at the ubiquinone site (Carelli et al., 1999). Interestingly, the G11778A LHON mutation decreases binding of rotenone, a complex I inhibitor that binds at or near the quinone redox site (Carelli et al., 1997). Furthermore, photoaffinity studies indicated that the ND1 subunit (site of the G3460A LHON mutation) also contributes to the rotenone binding region (Early et al., 1987). It has recently been shown that complex I contains a single, large domain that binds the various quinone-type inhibitors (Okun et al., 1999). Therefore, it is tempting to speculate that domains of the ND1, ND4 and ND6 subunits contribute to this inhibitor-binding domain and, by extrapolation, to the quinone redox site in the membrane-buried portion of the complex.
Six pathogenic mutations of the mtDNA ND6 gene have previously been associated with either LHON or LHON plus dystonia: G14459A/A72V (Jun et al., 1994; Shoffner et al., 1995), T14484C/M64V (Johns et al., 1992), C14482G/M64I (Howell et al., 1998), C14498T/Y59C (Wissinger et al., 1997), C14568T/G36S (Wissinger et al., 1997; Besch et al., 1999) and T14596A/I26M (De Vries et al., 1996). The results presented here, in conjunction with the previous studies, show that the 50-amino acid B/C domain of the ND6 subunit of complex I (amino acids 25–74, nucleotides 14452–14601) is a hot spot for LHON mutations (Figs 3 and 4) (Wissinger et al., 1997; Besch et al., 1999; Carelli et al., 1999).
Heteroplasmy and disease expression
Heteroplasmy is an uncommon finding in LHON pedigrees. Clinically affected G11778A LHON patients usually harbour >70% mutant mtDNA in their blood (Smith et al., 1993). It is therefore not surprising that the two affected males in the British LHON family and the affected Canadian male were found to have >80% mutant mtDNA in their blood. However, the affected British female had 51% mutant mtDNA in her blood (Fig. 1). This may be because a much higher level of mutant mtDNA was present in her optic nerve [as has been documented for the G11778A mutation (Howell et al., 1994)]. Alternatively, for an individual with a low level of mutant mtDNA, an additional serious physiological insult, such as coincidental vitamin B12 deficiency, may have precipitated the acute optic neuropathy. Finally, it is possible that the threshold level for the A14495G mutation might be lower than the threshold for other LHON mutations. It is particularly interesting to note that there was improvement in visual acuity in some of the affected A14495G LHON patients, because this is a common occurrence in LHON patients who carry the T14484C mutation in the ND6 gene (Nikoskelainen, 1994).
We have identified the seventh point mutation of the mtDNA ND6 gene as a cause of LHON. These observations add to the weight of evidence indicating the importance of the ND6 B/C hydrophobic domain in maintaining a healthy optic nerve throughout life. We suggest that direct sequencing of the B/C region of the ND6 gene should be carried out in all patients who present with a LHON-like optic neuropathy but who do not have the G11778A, G3460A or T14484C mutations.
Appendix I
Regions of the mitochondrial genome sequenced in the Canadian patient
The following mtDNA regions were cloned and sequenced in the Canadian patient in Galveston [numbered according to the rCRS (Anderson et al., 1981; Andrews et al., 1999)]: nucleotides 1–657; 2996–3250; 3286–3564; 4027–4294; 4689–4986; 6614–6904; 7365–7661; 9100–9386; 10 663–10 914; 10 865–11 313; 11 630–11 925; 12 562–12 848; 13 581–13 876; 13 846–14 138; 14 381–14 699; 14 576–14 872; 15 758–15 932; 15 909–16 569.
Appendix II
Primer extension assay for the A14495G mutation
A 500 base-pair PCR fragment encompassing nucleotide 14 495 was generated using the M13-tagged primers used for sequencing (L14227 forward M13 ccc ata atc ata caa agc cc; H14928 reverse M13 gtt gag gcg tct ggt gag) and AmpliTaq DNA polymerase (Perkin-Elmer), under the following conditions: 95°C denaturation for 5 min followed by 30 cycles of 95°C for 1 min, 58°C for 1 min and 72°C for 1 min, and final extension at 72°C for 10 min. Residual nucleotides in the PCR reaction mixtures were dephosphorylated [1 U of calf intestinal alkaline phosphatase (CAP), 5 μl of ×10 CAP buffer, incubated for 30 min at 37°C in a thermal cycler]. Following this, 1.1 μl of 0.25 M EDTA (pH 8.0) was added, and the CAP was denatured at 75°C for 10 min. The reaction products were purified down Qiaquick columns (QIAgen, Quiagen Hilden, Germany) and eluted in 30 μl of deionized water. One microlitre of the purified product was added to each 8-μl primer extension assay reaction, which included 0.64 U Thermo Sequenase DNA polymerase (Amersham), 25 μM dGTP and 25 μM ddATP (Roche), 10 mM Tris–HCl (pH 9.5), 5 mM KCl, 2 mM MgCl2, 0.002% Tween 20 and 20 fmol of 5′-FAM-labelled fluorescent primer (FAM aag aca acc atc att ccc cct; MWG Biotech AG, Ebersberg, Germany). The samples were denatured at 95°C for 2 min followed by 20 cycles of 95°C for 20 s and 55°C for 40 s. Two microlitres of each primer extension assay reaction was mixed with 1.5 μl deionized formamide, 0.5 μl ROX-350 size standard (Perkin-Elmer, Foster City, Calif., USA) and 1.5 μl of EDTA loading buffer (Perkin-Elmer), and denatured at 95°C for 5 min before immediate quenching on ice. The samples were analysed on an ABI 373 sequencer by electrophoresis in a 12% denaturing polyacrylamide gel. Raw data were analysed using Genescan v3.0 (ABI) and Genotyper v2.0 (ABI) software. The area under each curve was used to calculate the relative allele proportions.
Nucleotide changes from the revised Cambridge Reference Sequence* for the two families who harboured the A14495G/L60S mutation
| CRS position | rCRS nucleotide | LHON mtDNA sequences | |||||
|---|---|---|---|---|---|---|---|
| British LHON family | Canadian LHON family | ||||||
| Nucleotide | Gene† | Comment | Nucleotide | Gene† | Comment | ||
| n.t. = not tested (only part of the Canadian mtDNA was sequenced; see Patients and methods). *Cambridge Reference Sequence (Anderson et al., 1981); revised Cambridge Reference Sequence (R. M. Andrews et al., unpublished). Base substitutions are homoplasmic unless stated otherwise. †The number after the gene abbreviation refers to the amino acid position affected by the nucleotide substitution. For silent polymorphisms, we include the amino acid that is not changed as a result of the sequence change. ‡This polymorphism has been found to occur in a sub-branch of haplogroup H mtDNAs; the designation of polymorphisms as `common', `rare' and `private' is based primarily on our analysis of 60 complete European mtDNAs (R. M. Andrews et al., unpublished data), as well as on the appropriate published literature. | |||||||
| 152 | T | C | D-loop | Common polymorphism | T | – | – |
| 263 | A | G | D-loop | Common polymorphism | G | – | – |
| 750 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 1438 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 4216 | T | T | – | – | C | ND1/340 | Common polymorphism |
| Tyr→His | |||||||
| 4769 | A | G | ND2/100b | Common polymorphism | G | ND2/100 | Common polymorphism |
| Silent/Met | Silent/Met | ||||||
| 6776 | T | C | COX1/291 | H-specific polymorphism‡ | T | – | – |
| Silent/His | |||||||
| 8860 | A | G | ATPase 6/112 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 9196 | G | A | ATPase 6/224 | Private polymorphism | G | – | – |
| Asp→Asn | |||||||
| 10754 | A | C | ND4L/95 | Rare polymorphism | A | – | – |
| Silent/Leu | |||||||
| 11149 | G | G | – | – | A | ND4/130 | Private polymorphism |
| Heteroplasmic | Silent/Leu | ||||||
| 14470 | T | T | – | – | A | ND6/68 | Private polymorphism |
| Silent/Gly | |||||||
| 14495 | A | G | ND6/60 | Not previously described | G | ND6/60 | Not previously described |
| Heteroplasmic | Leu→Ser | Heteroplasmic | Leu→Ser | ||||
| 15326 | A | G | Cyt b/194 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 16390 | G | A | D-loop | Common polymorphism | G | – | – |
| 16519 | T | C | D-loop | Common polymorphism | C | – | – |
| CRS position | rCRS nucleotide | LHON mtDNA sequences | |||||
|---|---|---|---|---|---|---|---|
| British LHON family | Canadian LHON family | ||||||
| Nucleotide | Gene† | Comment | Nucleotide | Gene† | Comment | ||
| n.t. = not tested (only part of the Canadian mtDNA was sequenced; see Patients and methods). *Cambridge Reference Sequence (Anderson et al., 1981); revised Cambridge Reference Sequence (R. M. Andrews et al., unpublished). Base substitutions are homoplasmic unless stated otherwise. †The number after the gene abbreviation refers to the amino acid position affected by the nucleotide substitution. For silent polymorphisms, we include the amino acid that is not changed as a result of the sequence change. ‡This polymorphism has been found to occur in a sub-branch of haplogroup H mtDNAs; the designation of polymorphisms as `common', `rare' and `private' is based primarily on our analysis of 60 complete European mtDNAs (R. M. Andrews et al., unpublished data), as well as on the appropriate published literature. | |||||||
| 152 | T | C | D-loop | Common polymorphism | T | – | – |
| 263 | A | G | D-loop | Common polymorphism | G | – | – |
| 750 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 1438 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 4216 | T | T | – | – | C | ND1/340 | Common polymorphism |
| Tyr→His | |||||||
| 4769 | A | G | ND2/100b | Common polymorphism | G | ND2/100 | Common polymorphism |
| Silent/Met | Silent/Met | ||||||
| 6776 | T | C | COX1/291 | H-specific polymorphism‡ | T | – | – |
| Silent/His | |||||||
| 8860 | A | G | ATPase 6/112 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 9196 | G | A | ATPase 6/224 | Private polymorphism | G | – | – |
| Asp→Asn | |||||||
| 10754 | A | C | ND4L/95 | Rare polymorphism | A | – | – |
| Silent/Leu | |||||||
| 11149 | G | G | – | – | A | ND4/130 | Private polymorphism |
| Heteroplasmic | Silent/Leu | ||||||
| 14470 | T | T | – | – | A | ND6/68 | Private polymorphism |
| Silent/Gly | |||||||
| 14495 | A | G | ND6/60 | Not previously described | G | ND6/60 | Not previously described |
| Heteroplasmic | Leu→Ser | Heteroplasmic | Leu→Ser | ||||
| 15326 | A | G | Cyt b/194 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 16390 | G | A | D-loop | Common polymorphism | G | – | – |
| 16519 | T | C | D-loop | Common polymorphism | C | – | – |
Nucleotide changes from the revised Cambridge Reference Sequence* for the two families who harboured the A14495G/L60S mutation
| CRS position | rCRS nucleotide | LHON mtDNA sequences | |||||
|---|---|---|---|---|---|---|---|
| British LHON family | Canadian LHON family | ||||||
| Nucleotide | Gene† | Comment | Nucleotide | Gene† | Comment | ||
| n.t. = not tested (only part of the Canadian mtDNA was sequenced; see Patients and methods). *Cambridge Reference Sequence (Anderson et al., 1981); revised Cambridge Reference Sequence (R. M. Andrews et al., unpublished). Base substitutions are homoplasmic unless stated otherwise. †The number after the gene abbreviation refers to the amino acid position affected by the nucleotide substitution. For silent polymorphisms, we include the amino acid that is not changed as a result of the sequence change. ‡This polymorphism has been found to occur in a sub-branch of haplogroup H mtDNAs; the designation of polymorphisms as `common', `rare' and `private' is based primarily on our analysis of 60 complete European mtDNAs (R. M. Andrews et al., unpublished data), as well as on the appropriate published literature. | |||||||
| 152 | T | C | D-loop | Common polymorphism | T | – | – |
| 263 | A | G | D-loop | Common polymorphism | G | – | – |
| 750 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 1438 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 4216 | T | T | – | – | C | ND1/340 | Common polymorphism |
| Tyr→His | |||||||
| 4769 | A | G | ND2/100b | Common polymorphism | G | ND2/100 | Common polymorphism |
| Silent/Met | Silent/Met | ||||||
| 6776 | T | C | COX1/291 | H-specific polymorphism‡ | T | – | – |
| Silent/His | |||||||
| 8860 | A | G | ATPase 6/112 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 9196 | G | A | ATPase 6/224 | Private polymorphism | G | – | – |
| Asp→Asn | |||||||
| 10754 | A | C | ND4L/95 | Rare polymorphism | A | – | – |
| Silent/Leu | |||||||
| 11149 | G | G | – | – | A | ND4/130 | Private polymorphism |
| Heteroplasmic | Silent/Leu | ||||||
| 14470 | T | T | – | – | A | ND6/68 | Private polymorphism |
| Silent/Gly | |||||||
| 14495 | A | G | ND6/60 | Not previously described | G | ND6/60 | Not previously described |
| Heteroplasmic | Leu→Ser | Heteroplasmic | Leu→Ser | ||||
| 15326 | A | G | Cyt b/194 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 16390 | G | A | D-loop | Common polymorphism | G | – | – |
| 16519 | T | C | D-loop | Common polymorphism | C | – | – |
| CRS position | rCRS nucleotide | LHON mtDNA sequences | |||||
|---|---|---|---|---|---|---|---|
| British LHON family | Canadian LHON family | ||||||
| Nucleotide | Gene† | Comment | Nucleotide | Gene† | Comment | ||
| n.t. = not tested (only part of the Canadian mtDNA was sequenced; see Patients and methods). *Cambridge Reference Sequence (Anderson et al., 1981); revised Cambridge Reference Sequence (R. M. Andrews et al., unpublished). Base substitutions are homoplasmic unless stated otherwise. †The number after the gene abbreviation refers to the amino acid position affected by the nucleotide substitution. For silent polymorphisms, we include the amino acid that is not changed as a result of the sequence change. ‡This polymorphism has been found to occur in a sub-branch of haplogroup H mtDNAs; the designation of polymorphisms as `common', `rare' and `private' is based primarily on our analysis of 60 complete European mtDNAs (R. M. Andrews et al., unpublished data), as well as on the appropriate published literature. | |||||||
| 152 | T | C | D-loop | Common polymorphism | T | – | – |
| 263 | A | G | D-loop | Common polymorphism | G | – | – |
| 750 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 1438 | A | G | 12sRNA | Common polymorphism | n.t. | n.t. | n.t. |
| 4216 | T | T | – | – | C | ND1/340 | Common polymorphism |
| Tyr→His | |||||||
| 4769 | A | G | ND2/100b | Common polymorphism | G | ND2/100 | Common polymorphism |
| Silent/Met | Silent/Met | ||||||
| 6776 | T | C | COX1/291 | H-specific polymorphism‡ | T | – | – |
| Silent/His | |||||||
| 8860 | A | G | ATPase 6/112 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 9196 | G | A | ATPase 6/224 | Private polymorphism | G | – | – |
| Asp→Asn | |||||||
| 10754 | A | C | ND4L/95 | Rare polymorphism | A | – | – |
| Silent/Leu | |||||||
| 11149 | G | G | – | – | A | ND4/130 | Private polymorphism |
| Heteroplasmic | Silent/Leu | ||||||
| 14470 | T | T | – | – | A | ND6/68 | Private polymorphism |
| Silent/Gly | |||||||
| 14495 | A | G | ND6/60 | Not previously described | G | ND6/60 | Not previously described |
| Heteroplasmic | Leu→Ser | Heteroplasmic | Leu→Ser | ||||
| 15326 | A | G | Cyt b/194 | Common polymorphism | n.t. | n.t. | n.t. |
| Thr→Ala | |||||||
| 16390 | G | A | D-loop | Common polymorphism | G | – | – |
| 16519 | T | C | D-loop | Common polymorphism | C | – | – |
Sequence variation of the 35 amino acid hydrophobic ND6 B/C transmembrane domain (amino acid residues 25–74 inclusive) in 271 healthy and disease controls
| ND6 gene polymorphism | ND6 protein | Epidemiology | ||||
|---|---|---|---|---|---|---|
| CRS position | rCRS nucleotide | Observed polymorphism | Position | Amino acid change | Frequency | Clinical status |
| CRS = Cambridge Reference Sequence (Anderson et al., 1981); rCRS = revised Cambridge Reference Sequence (Andrews et al., 1999). The disease control group does not include patients who are known to harbour the T14484C primary LHON mutation. | ||||||
| 14 470 | T | C | 68 | Silent | 4 | Various, including normals |
| 14 497 | A | G | 59 | Silent | 1 | Normal |
| 14 551 | T | C | 41 | Silent | 1 | Normal |
| 14 560 | G | A | 38 | Silent | 4 | Various, including normals |
| 14 566 | A | G | 36 | Silent | 8 | Various, including normals |
| 14 569 | G | A | 35 | Silent | 2 | 11778 LHON |
| 14 572 | A | G | 34 | Silent | 1 | Kearns–Sayre syndrome |
| 14 577 | T | C | 33 | I→V | 1 | Normal |
| 14 582 | A | G | 31 | V→A | 1 | Normal |
| 14 587 | A | G | 29 | Silent | 1 | Normal |
| ND6 gene polymorphism | ND6 protein | Epidemiology | ||||
|---|---|---|---|---|---|---|
| CRS position | rCRS nucleotide | Observed polymorphism | Position | Amino acid change | Frequency | Clinical status |
| CRS = Cambridge Reference Sequence (Anderson et al., 1981); rCRS = revised Cambridge Reference Sequence (Andrews et al., 1999). The disease control group does not include patients who are known to harbour the T14484C primary LHON mutation. | ||||||
| 14 470 | T | C | 68 | Silent | 4 | Various, including normals |
| 14 497 | A | G | 59 | Silent | 1 | Normal |
| 14 551 | T | C | 41 | Silent | 1 | Normal |
| 14 560 | G | A | 38 | Silent | 4 | Various, including normals |
| 14 566 | A | G | 36 | Silent | 8 | Various, including normals |
| 14 569 | G | A | 35 | Silent | 2 | 11778 LHON |
| 14 572 | A | G | 34 | Silent | 1 | Kearns–Sayre syndrome |
| 14 577 | T | C | 33 | I→V | 1 | Normal |
| 14 582 | A | G | 31 | V→A | 1 | Normal |
| 14 587 | A | G | 29 | Silent | 1 | Normal |
Sequence variation of the 35 amino acid hydrophobic ND6 B/C transmembrane domain (amino acid residues 25–74 inclusive) in 271 healthy and disease controls
| ND6 gene polymorphism | ND6 protein | Epidemiology | ||||
|---|---|---|---|---|---|---|
| CRS position | rCRS nucleotide | Observed polymorphism | Position | Amino acid change | Frequency | Clinical status |
| CRS = Cambridge Reference Sequence (Anderson et al., 1981); rCRS = revised Cambridge Reference Sequence (Andrews et al., 1999). The disease control group does not include patients who are known to harbour the T14484C primary LHON mutation. | ||||||
| 14 470 | T | C | 68 | Silent | 4 | Various, including normals |
| 14 497 | A | G | 59 | Silent | 1 | Normal |
| 14 551 | T | C | 41 | Silent | 1 | Normal |
| 14 560 | G | A | 38 | Silent | 4 | Various, including normals |
| 14 566 | A | G | 36 | Silent | 8 | Various, including normals |
| 14 569 | G | A | 35 | Silent | 2 | 11778 LHON |
| 14 572 | A | G | 34 | Silent | 1 | Kearns–Sayre syndrome |
| 14 577 | T | C | 33 | I→V | 1 | Normal |
| 14 582 | A | G | 31 | V→A | 1 | Normal |
| 14 587 | A | G | 29 | Silent | 1 | Normal |
| ND6 gene polymorphism | ND6 protein | Epidemiology | ||||
|---|---|---|---|---|---|---|
| CRS position | rCRS nucleotide | Observed polymorphism | Position | Amino acid change | Frequency | Clinical status |
| CRS = Cambridge Reference Sequence (Anderson et al., 1981); rCRS = revised Cambridge Reference Sequence (Andrews et al., 1999). The disease control group does not include patients who are known to harbour the T14484C primary LHON mutation. | ||||||
| 14 470 | T | C | 68 | Silent | 4 | Various, including normals |
| 14 497 | A | G | 59 | Silent | 1 | Normal |
| 14 551 | T | C | 41 | Silent | 1 | Normal |
| 14 560 | G | A | 38 | Silent | 4 | Various, including normals |
| 14 566 | A | G | 36 | Silent | 8 | Various, including normals |
| 14 569 | G | A | 35 | Silent | 2 | 11778 LHON |
| 14 572 | A | G | 34 | Silent | 1 | Kearns–Sayre syndrome |
| 14 577 | T | C | 33 | I→V | 1 | Normal |
| 14 582 | A | G | 31 | V→A | 1 | Normal |
| 14 587 | A | G | 29 | Silent | 1 | Normal |
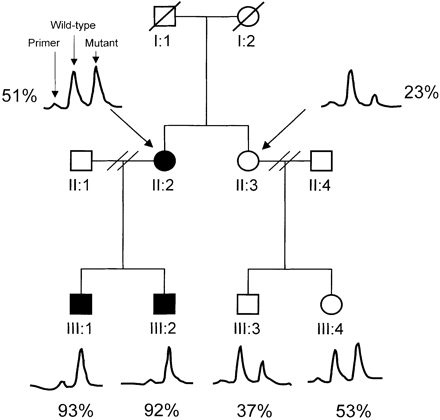
Pedigree of the British LHON family. The percentage of the A14495G mutation in blood, as determined by primer extension assay, in each individual is indicated. Solid symbols indicate individuals with a clinical diagnosis of LHON. Tracings of the primer extension assay results are included (for experimental details, see Patients and methods and Appendix II).
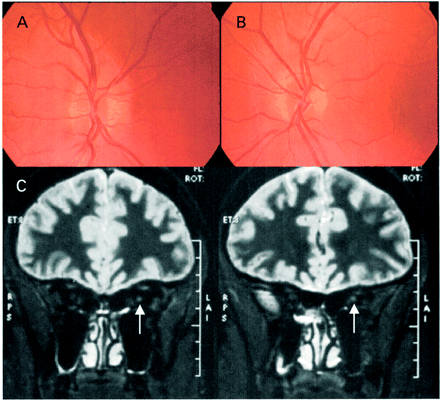
Acute fundal appearance (A, right; B, left) and T2-weighted coronal MRI images (C) of III-2 from the British LHON family. The arrow indicates the high-signal region in the left optic nerve.
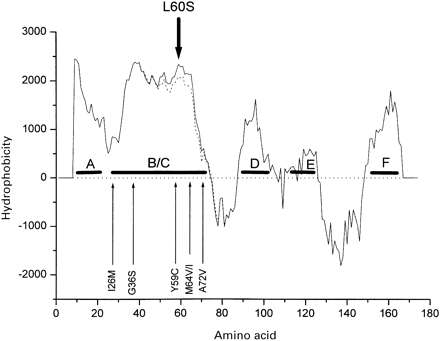
Hydropathy plot for human ND6. Hydropathy analysis of the human ND6 gene using the Kyte–Doolittle algorithm. The putative hydrophobic transmembrane helices are lettered A to E. The position of known point mutations affecting the ND6 gene are indicated by the vertical arrows that show their position in the amino acid sequence. G14459A/A72V (Jun et al., 1994; Shoffner et al., 1995), T14484C/M64V (Johns et al., 1992), C14482G/M64I (Howell et al., 1998), C14498T/Y59C (Wissinger et al., 1997), C14568T/G36S (Wissinger et al., 1997; Besch et al., 1999) and T14596A/I26M (De Vries et al., 1996) are previously published pathogenic mutations. The wild-type ND6 structure (solid line) is altered by the presence of the A14495G/L60S mutation (dotted line). Most of the pathogenic mutations in this region have minor effects on the predicted protein structure.
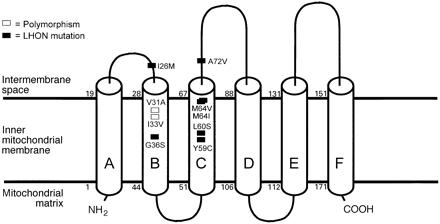
Predicted transmembrane structure of human ND6. The proposed transmembrane helical structure of the ND6 protein as predicted with the TMpred program (see text). Cylinders represent transmembrane helices. In addition to the A14495G/L60S reported here, six established pathogenic mutations associated with LHON, with or without additional abnormalities including dystonia, are shown together with the benign substitution polymorphisms within the B and C transmembrane helices. G14459A/A72V (Jun et al., 1994; Shoffner et al., 1995), T14484C/M64V (Johns et al., 1992), C14482G/M64I (Howell et al., 1998), C14498T/Y59C(Wissinger et al., 1997), C14568T/G36S (Wissinger et al., 1997; Besch et al., 1999) and T14596A/I26M (De Vries et al., 1996) are previously published pathogenic mutations. A14577G/I33V and A14582G/V31A are polymorphisms that have not been reported previously.
We wish to thank Dr E. Fahy (MitoKor, San Diego, Calif., USA) for his assistance in setting up the primer extension reaction. P.F.C. is supported by the Wellcome Trust and N.H. is supported by the Eierman Foundation and the National Eye Institute (RO1 EY10758).
References
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome.
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge Reference Sequence for human mitochondrial DNA [letter].
Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme.
Besch D, Leo-Kottler B, Zrenner E, Wissinger B. Leber's hereditary optic neuropathy: clinical and molecular genetic findings in a patient with a new mutation in the ND6 gene.
Brown MD. The enigmatic relationship between mitochondrial dysfunction and Leber's hereditary optic neuropathy [editorial]. [Review].
Carelli V, Ghelli A, Ratta M, Bacchilega E, Sangiorgi S, Mancini R, et al. Leber's hereditary optic neuropathy: biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype.
Carelli V, Ghelli A, Bucchi L, Montagna P, De Negri A, Leuzzi V, et al. Biochemical features of mtDNA 14484 (ND6/M64V) point mutation associated with Leber's hereditary optic neuropathy.
Chinnery PF, Howell N, Andrews RM, Turnbull DM. Mitochondrial DNA analysis: polymorphisms and pathogenicity. [Review].
Cock HR, Cooper JM, Schapira AH. The 14484 ND6 mtDNA mutation in Leber hereditary optic neuropathy does not affect fibroblast complex I activity [letter].
De Vries DD, Went LN, Bruyn GW, Scholte HR, Hofstra RM, Bolhuis PA, et al. Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia.
Early FG, Patel SD, Ragan I, Attardi G. Photolabelling of mitochondrially encoded subunit of NADH dehydrogenase with [3H]dihydrorotenone.
Fahy E, Nazarbaghi R, Zomorrodi M, Herrnstadt C, Parker WD, Davis RE, et al. Multiplex fluorescence-based primer extension method for quantitative mutation analysis of mitochondrial DNA and its diagnostic application for Alzheimer's disease.
Fearnley IM, Walker JE. Conservation of sequences of subunits of mitochondrial complex I and their relationships with other proteins. [Review].
Friedrich T, Strohdeicher M, Hofhaus G, Preis D, Sahm H, Weiss H. The same domain motif for ubiquinone reduction in mitochondrial or chloroplast NADH dehydrogenase and bacterial glucose dehydrogenase.
Ghosh SS, Fahy E, Bodis-Wollner I, Sherman J, Howell N. Longitudinal study of a heteroplasmic 3460 Leber hereditary optic neuropathy family by multiplexed primer-extension analysis and nucleotide sequencing.
Grigorieff N. Three-dimensional structure of bovine NADH:ubiquinone oxidoreductase (complex I) at 22 A in ice.
Harding AE, Sweeney MG, Govan GG, Riordan-Eva P. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation.
Howell N. Mitochondrial gene mutations and human diseases: a prolegomenon [editorial].
Howell N. Leber hereditary optic neuropathy: how do mitochondrial DNA mutations cause degeneration of the optic nerve? [Review].
Howell N, Bindoff LA, McCullough DA, Kubacka I, Poulton J, Mackey D, et al. Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees.
Howell N, Xu M, Halvorson S, Bodis-Wollner I, Sherman J. A heteroplasmic LHON family: tissue distribution and transmission of the 11778 mutation [letter].
Howell N, Kubacka I, Halvorson S, Howell B, McCullough DA, Mackey D. Phylogenetic analysis of the mitochondrial genomes from Leber hereditary optic neuropathy pedigrees.
Howell N, Bogolin C, Jamieson R, Marenda DR, Mackey DA. mtDNA mutations that cause optic neuropathy: how do we know? [letter].
Johns DR, Berman J. Alternative, simultaneous complex I mitochondrial DNA mutations in Leber's hereditary optic neuropathy.
Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy.
Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia.
Lanave C, Attimonelli M, De Robertis M, Licciulli F, Liuni S, Sbisa E, et al. Update of AMmtDB: a database of multi-aligned metazoa mitochondrial DNA sequences.
Larsson N-G, Andersen O, Holme E, Oldfors A, Wahlstrom J. Leber's hereditary optic neuropathy and complex I deficiency in muscle.
Leber T. Uber hereditäre und congenital–angelegte Sehnervenleiden.
Lodi R, Taylor DJ, Tabrizi SJ, Kumar S, Sweeney M, Wood NW, et al. In vivo skeletal muscle mitochondrial function in Leber's hereditary optic neuropathy assessed by 31P magnetic resonance spectroscopy.
Mackey D, Howell N. A variant of Leber hereditary optic neuropathy characterized by recovery of vision and by an unusual mitochondrial genetic etiology.
Mackey DA, Oostra R-J, Rosenberg T, Nikoskelainen E, Bronte-Stewart J, Poulton J, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy [letter].
Macmillan C, Kirkham T, Fu K, Allison V, Andermann E, Chitayat D, et al. Pedigree analysis of French Canadian families with the T14484C Leber's hereditary optic neuropathy.
Nicholas KB, Nicholas HB. Genedoc: a tool for editing and annotating multiple sequence alignments. Distributed by the author, 1997.
Okun JG, Lummen P, Brandt U. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase).
Ozawa T, Tanaka M, Sugiyama S, Ino H, Ohno K, Hattori K, et al. Patients with idiopathic cardiomyopathy belong to the same mitochondrial DNA gene family of Parkinson's disease and mitochondrial encephalomyopathy.
Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical features of Leber's hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation.
Shoffner JM, Brown MD, Stugard C, Jun AS, Pollock S, Haas RH, et al. Leber's hereditary optic neuropathy plus dystonia is caused by a mitochondrial DNA point mutation.
Smeitink J, van den Heuvel L. Human mitochondrial complex I in health and disease. [Review].
Smith KH, Johns DR, Heher KL, Miller NR. Heteroplasmy in Leber's hereditary optic neuropathy.
Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, et al. Classification of European mtDNAs from an analysis of three European populations.
Walker JE. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. [Review].
Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy.


{kind=link}
{kind=link}
{kind=link}
{kind=link}