




Abstract
Motor practice may lead to expansion of trained representations in the motor cortex, but it is unknown whether this practice-dependent plasticity can be purposefully enhanced or depressed. Evidence, mainly based on animal experiments, indicates that the activity of GABA-related cortical inhibition is important in controlling the extent to which plasticity may occur. We tested the role of GABA in modulating practice-dependent plasticity in the human motor cortex. A decrease in GABA-related cortical inhibition was achieved by ischaemic nerve block (INB) in the hand by deafferentation/deefferentation and an increase was achieved by administration of the GABAA receptor agonist lorazepam. In Experiment 1, healthy subjects performed motor practice (MP), consisting of repeated ballistic contractions of the biceps muscle in the absence (MP alone) or presence of INB (MP+INB). Changes in the biceps motor cortex representation were assessed by transcranial magnetic stimulation (TMS). MP+INB resulted in a dramatic increase in the size of the motor evoked potential (MEP) and in paired-pulse excitability compared with mild or no changes in the MP-alone and INB-alone conditions. In Experiment 2, this dramatic increase in biceps representation induced by MP+INB was replicated when subjects were pretreated with placebo, but this increase was prevented or even switched to a decrease when subjects were pretreated with lorazepam. These findings indicate that a decrease in GABA-related inhibition facilitates practice-dependent plasticity in the human motor cortex, whereas an increase depresses it. In Experiment 3, practice-dependent plasticity (assessed by TMS, as in the first two experiments) was also tested at the behavioural level. The dramatic increase in biceps MEP size induced by MP+INB was paralleled by an increase in peak acceleration of the fastest elbow flexion movements. Similarly, the lack of change in MEP size in the MP-alone condition was paralleled by a lack of change in peak acceleration. We propose that changes in GABA activity may be instrumented to modulate plasticity purposefully; for instance, to enhance plastic change and recovery of function after a lesion in neurological patients.
Introduction
The last two decades have provided ample evidence that the adult non-human and human primate sensorimotor cortex maintains the capacity for plastic change following lesions (for reviews, see Merzenich and Kaas, 1982; Kaas et al., 1983; Kaas, 1991) or in the process of learning (for reviews, see Weinberger, 1995; Donoghue et al., 1996; Sanes and Donoghue, 2000). Some forms of plasticity can occur rapidly, within minutes to hours. In the somatosensory cortex, neurones deafferented by peripheral nerve lesion or amputation rapidly become responsive to sensory input from adjacent intact body sites (Merzenich et al., 1983; Kolarik et al., 1994; Silva et al., 1996; Tinazzi et al., 1997; Borsook et al., 1998). In the motor cortex, representations can reorganize rapidly in response to peripheral nerve lesion or ischaemic nerve block (Sanes et al., 1988; Donoghue et al., 1990; Brasil-Neto et al., 1992, 1993; Ridding and Rothwell, 1995; Ziemann et al., 1998c), or during motor practice (Pascual-Leone et al., 1993, 1995b; Nudo et al., 1996a, b; Classen et al., 1998; Bütefisch et al., 2000) and motor learning (Pascual-Leone et al., 1994, 1995a). Two main mechanisms have been proposed to account for this rapid plasticity. One is the unmasking of latent horizontal connections (for reviews, see Sanes and Donoghue, 1997, 2000) and the other is modification of the strength of synaptic contacts, such as by long-term potentiation (LTP) and long-term depression (LTD) (for review, see Hess and Donoghue, 1996b). To some extent, both concepts rely on the view that the motor cortex is a dynamic substrate that contains multiple, overlapping motor representations (Donoghue et al., 1992; Wassermann et al., 1992; Schieber and Hibbard, 1993; Rao et al., 1995; Sanes et al., 1995) and a network of extensive horizontal connections (Huntley and Jones, 1991). Several experiments have provided direct evidence that in the motor cortex both unmasking (Huntley, 1997) and LTP/LTD (Hess and Donoghue, 1994, 1996a) are mediated, and constrained, by the pre-existing horizontal connectivity. Another important property of unmasking and LTP/LTD is that they require or are significantly enhanced by a reduction in local inhibition (Jacobs and Donoghue, 1991; Hess and Donoghue, 1994; Hess et al., 1996).
Magnetic resonance spectroscopy experiments in humans have shown a rapid decrease in GABA in the sensorimotor cortex contralateral to transient ischaemic deafferentation of the hand (Levy et al., 1999) and in the visual cortex after light deprivation (Boroojerdi et al., 2000). Previously, we have shown that during deafferentation-induced disinhibition in the sensorimotor cortex the effects of repetitive TMS (rTMS) are dramatically enhanced (Ziemann et al., 1998b). In particular, slow-rate rTMS, which was ineffective in producing changes in motor output when given alone (without deafferentation), resulted in a dramatic and long-lasting increase in motor output to the biceps muscle when given during deafferentation of the hand. Here, we tested (Experiment 1) whether the effects of motor practice are also enhanced when motor practice is performed during motor cortex disinhibition.
Studies of the rat neocortex have shown that LTP switches to LTD as a function of decreasing postsynaptic excitability (Artola et al., 1990; Bear and Kirkwood, 1996). Therefore, we hypothesized that practice-dependent plasticity may be depressed if motor practice is performed during increased cortical inhibition. We tested this hypothesis (Experiment 2) by having subjects perform the motor practice during hand deafferentation after having been pretreated with the GABAA receptor agonist lorazepam. Together, Experiments 1 and 2 were designed to assess the role of GABA-related cortical inhibition in modifying practice-dependent plasticity.
In both experiments, practice-dependent changes in motor cortical output to the practice muscle (the biceps brachii muscle) were studied with TMS, which is an established means to assess motor cortical plasticity (Cohen et al., 1998). Motor threshold (MT), motor evoked potential (MEP) amplitude and paired-pulse excitability (PPE) were used to test the membrane-related excitability of corticocortical axons (Ziemann et al., 1996b), corticospinal excitability (Devanne et al., 1997) and the synaptic efficacy of inhibitory and excitatory circuitry at the level of the motor cortex (Kujirai et al., 1993; Ziemann et al., 1996c), respectively.
Finally, to link these TMS measures of cortical plasticity with motor performance, we evaluated practice-dependent changes in the kinematics of the fastest voluntary elbow flexion movements in addition to the changes in TMS-evoked motor cortical output (Experiment 3). We hypothesized that the kinematics of ballistic movement changes in parallel with TMS-evoked motor output, because ballistic movements are generated through activity in the motor cortex and the fastest-conducting corticospinal neurones (Fromm and Evarts, 1981). Furthermore, ballistic activity is closely linked to TMS measures of motor excitability (Mills and Kimiskidis, 1996).
Methods
Subjects
In each of the three experiments (see below), six different healthy, right-handed subjects were investigated (mean age 25.7 ± 4.2, 35.7 ± 8.7 and 33.5 ± 9.4 years, respectively). All experiments were approved by the Institutional Review Board of the National Institute of Neurological Disorders and Stroke and were conducted according to the Declaration of Helsinki. All subjects gave their written informed consent.
Experiment 1
Interventions
Five interventions were tested in each subject in separate sessions conducted at least 1 week apart: ischaemic nerve block (INB) alone to induce motor cortex disinhibition (Levy et al., 1999); motor practice alone (MP); motor practice during INB (MP+INB); electrical stimulation (ES) of the biceps during INB (ES+INB); and rapid passive movements (PAS) of the elbow during INB (PAS+INB). For INB, a pneumatic tourniquet was placed distal to the left elbow and inflated to 220–250 mmHg for an average duration of 45 min. For MP, subjects performed repeated (rate 0.1 Hz), externally paced (by an auditory `go' signal) voluntary elbow flexion movements by briefly contracting the left biceps in a twitch-like fashion. ES (0.2 ms square-wave constant-current pulses, cathode on motor point of left biceps, average muscle twitch amplitude 0.57 ± 0.22 mV) and rapid PAS of the left elbow (accomplished by the experimenter) were applied at the rate of 0.1 Hz while the subject did not practice. MP, ES and PAS started at the time of tourniquet inflation and were discontinued on reaching complete motor nerve block, defined as the time when motor responses in a hand muscle, the abductor pollicis brevis (APB), were no longer elicited by TMS (mean 31.1 min).
MP and MP+INB defined the major comparison for testing the extent to which the effects of MP on motor cortex output was enhanced by INB-induced disinhibition. INB was an important control experiment in order to quantify the changes induced by ischaemic forearm deafferentation alone (Ziemann et al., 1998b). Finally, ES+INB and PAS+INB mimicked afferent signals produced by MP (muscle twitch and elbow joint movement, respectively). Therefore, comparison of these control experiments with MP+INB tested the relative contributions of afferent input and voluntary motor cortex activation to the changes in motor cortex output induced by MP+INB.
Measurements
TMS-evoked motor cortical output was measured by surface electromyography (EMG) (bandpass 0.1–2.5 kHz) from the left APB and the left biceps muscle, using Ag–AgCl cup electrodes in a belly–tendon montage and a Counterpoint Electromyograph (Dantec Electronics, Skovlunde, Denmark). The raw EMG was digitized at a rate of 5 kHz and stored on an IBM 486 AT-compatible laboratory computer for off-line analysis. A figure-of-eight-shaped stimulating coil connected to a Bistim module (Magstim, Whitland, Dyfed, UK) was positioned on the scalp over the right motor cortex at the optimal site for eliciting MEPs in the left biceps. MT was determined to the nearest 1% of the maximum stimulator output and defined as the minimum stimulus intensity to evoke MEPs of ≥50 μV in at least five of 10 trials with the biceps at rest (Rossini et al., 1994). Peak-to-peak MEP amplitude was measured at stimulus intensities of 20 and 30% of stimulator output above the biceps MT (five trials each). PPE was tested in a conditioning–test stimulus paradigm (Kujirai et al., 1993; Ziemann et al., 1996c). The intensity of the test stimulus was adjusted to produce a control MEP of 200–500 μV when given alone. The conditioning stimulus was set to 80% of the MT in the APB. Such low-intensity TMS does not produce significant corticospinal activation (Di Lazzaro et al., 1998). Therefore, any effect of the conditioning stimulus on the control MEP was attributable to intracortical mechanisms. PPE was tested at interstimulus intervals of 4, 10 and 15 ms by presenting eight trials for each of the three intervals and eight control trials in pseudorandomized order. For each interval, the mean conditioned MEP was expressed as a percentage of the control mean. As can be seen from Fig. 1C, the conditioning–test interval of 4 ms resulted in inhibition of the test MEP (before intervention across all interventions, 66.9 ± 24.2%) while the intervals of 10 and 15 ms resulted in facilitation (153.1 ± 74.9 and 162.6 ± 63.4%, respectively). Because the statistical analysis (see below) did not show an effect of interstimulus interval, the three intervals were pooled to one variable (PPE) and were not treated separately. All measurements were made with the biceps at rest, monitored by continuous audiovisual feedback of the biceps EMG. The measures were obtained before intervention (pre-measurement), at the end of intervention, i.e. 5 min after completion of INB or motor practice, and 20, 40 and 60 min later (post-measurements).
In order to quantify the voluntary EMG activity of the ballistic biceps contractions during MP and MP+INB, the EMG was recorded through the same surface electrodes as those used for the MEP measurements. The EMG was single-trial-rectified, aligned to the onset of the EMG burst and then averaged across all recorded trials (mean, 186 trials). The average was smoothed with a gliding sledge that averaged across 10 neighbouring data points (2 ms). From this rectified and smoothed signal of the voluntary biceps EMG burst, the peak amplitude (in mV) and the 25–75% onset-to-peak rise time (in milliseconds) were measured.
Experiment 2
Interventions
Subjects performed MP+INB (as in Experiment 1), once after pretreatment with a single oral dose of 2 mg of the GABAA receptor agonist lorazepam [7-chlor-5-(2-chlorphenyl)-3- hydroxy-1H-1,4-benzodiazepin-2(3H)-on], a short-acting benzodiazepine, and a second time (1 week later) after pretreatment with placebo. The order of drugs was balanced across subjects. The drugs were administered 2.5 h before the start of the measurements. In all subjects, lorazepam induced mild sedation, which did not interfere with the subjects' ability to comply fully with the requirements of the practice task.
Measurements
TMS-evoked motor cortical output was measured the same way as in Experiment 1.
Experiment 3
Interventions
Subjects performed MP and MP+INB in two separate sessions, as described for Experiment 1.
Measurements
TMS-evoked motor output was measured as MEP amplitude in the biceps muscle, as described above. In addition, voluntary motor output was measured as the 10–90% rise time and the peak of the acceleration signal produced by maximum ballistic elbow flexion movements, using a piezoelectric accelerometer (Endevco, San Juan Capistrano, Calif., USA) attached to the volar surface of the forearm. The subject sat upright in a chair with the forearm supinated and the elbow flexed at 90° and fixed to the armrest. The ballistic movements were made in the vertical plane in response to an auditory `go' signal; there were 15 trials each at 0.1 Hz before intervention (pre-measurement) and 10, 30 and 50 min after intervention (post-measurements). Subjects were instructed to perform full-range elbow flexion movements, i.e. from the starting position of 90° between forearm and upper arm to maximal elbow flexion. Trials were discarded from off-line analysis if the flexion movement stopped short. To avoid possible carry-over effects between the two experiments (MP versus MP+INB), we studied the left arm of a given subject in one experiment and the right arm in the other experiment. The orders of arm and intervention were pseudorandomized and balanced across subjects.
Statistics
Each measure (M) was analysed separately. For each time point, changes were expressed as (Mpost – Mpre)/Mpre. The within-subject factors of intervention and time were evaluated with a repeated-measures analysis of variance. The levels of M (two stimulus intensities for MEP amplitude, three interstimulus intervals for PPE) were also tested but never showed a significant effect. Therefore, the data reported here are the averages of MEP amplitude and PPE across levels. Post hoc paired comparisons were performed with Fisher's protected least significant difference multiple t statistic. The significance level was defined as P < 0.05.
Results
Experiment 1
MP+INB resulted in a significantly greater increase in MEP amplitude of the biceps muscle than MP alone, INB alone (no practice) or proprioceptive feedback during deafferentation (no practice), mimicked by electrical stimulation of the biceps muscle (ES+INB) or passive elbow movements (PAS+INB) (P < 0.05; Fig. 1A and D). This enhancement was not explained simply by an additive effect because the increase in MEP amplitude produced by MP+INB was significantly larger than the algebraic sum of the increases produced by MP alone and INB alone (2.61 ± 1.27 versus 1.14 ± 1.06, P = 0.034) (Fig. 1A, measurements made late in the intervention period). Furthermore, MP+INB was the only intervention to induce a significant increase in PPE (P < 0.05) (Fig. 1C and D). This was due to both a slight reduction in inhibition at the interstimulus interval of 4 ms and a clear increase in facilitation at the intervals of 10 and 15 ms (Fig. 1C). MT was unaffected by intervention.
MP was performed the same way with and without INB. This was inferred from the monitoring of the rectified and smoothed EMG of the voluntary biceps burst during MP, which revealed no difference with and without INB (mean peak amplitude 0.24 ± 0.15 versus 0.26 ± 0.10 mV; mean 25–75% rise time 20.0 ± 7.1 versus 24.7 ± 5.9 ms; P = 0.68 and 0.18, respectively).
Furthermore, there were no differences between interventions in the measures of TMS-evoked motor cortical output to the biceps before the start of interventions (premeasurements) or in the duration of intervention (P = 0.20–0.98). Therefore, these factors can be excluded as explanations of the INB-induced enhancement of practice-dependent plasticity.
Experiment 2
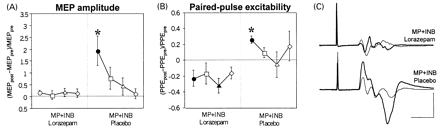
After pretreatment of the subjects with placebo, MP+INB resulted in a strong increase in biceps MEP amplitude, replicating the findings of Experiment 1, whereas no increase in MEP amplitude occurred when subjects were pretreated with lorazepam (Fig. 2A and C). PPE increased late into INB when subjects were pretreated with placebo, but decreased in subjects who had received lorazepam (Fig. 2B). MT was unaffected by intervention.
The EMG of the voluntary biceps burst did not show a significant difference during MP in the lorazepam versus placebo condition (mean peak amplitude 0.32 ± 0.18 versus 0.34 ± 0.18 mV; mean 25–75% rise time 21.9 ± 4.1 versus 23.2 ± 5.9 ms; P = 0.47 and 0.57, respectively).
Furthermore, there were no differences between conditions in the measures of TMS-evoked motor output to the biceps before the start of interventions (premeasurements) or in the duration of intervention (P = 0.12–0.55).
Experiment 3
MP+INB resulted in an increase in the mean peak amplitude of the voluntary EMG burst (before intervention and 10, 30 and 50 min after intervention: 0.63 ± 0.31, 0.74 ± 0.36 0.65 ± 0.28 and 0.77 ± 0.36 mV, respectively; for normalized data see Fig. 3B), whereas this increase was absent after MP alone (mean amplitudes before and 10, 30 and 50 min after intervention: 0.78 ± 0.25, 0.79 ± 0.24, 0.79 ± 0.18 and 0.80 ± 0.13 mV, respectively; cf. Fig. 3A). This difference was matched by a significantly stronger increase in peak acceleration and a significantly stronger decrease in the 10–90% rise time of the acceleration signal after MP+INB when compared with the non-significant changes produced by MP alone (Fig. 3C and D). This practice-dependent improvement in movement kinematics during INB was accompanied by an increase in MEP amplitude in the biceps muscle in each subject (Fig. 3E and F).
During motor practice, monitoring of the EMG of the voluntary biceps burst did not show a significant difference between the MP alone and the MP+INB condition (mean peak amplitude 0.59 ± 0.09 versus 0.37 ± 0.16 mV; mean 25–75% rise time 18.0 ± 2.9 versus 21.9 ± 8.6 ms; P = 0.09 and 0.54, respectively), indicating that the motor practice was performed the same way across the two interventions. There were also no differences in MEP amplitude and movement kinematics before the start of intervention (pre-measurements) or in the duration of intervention (P = 0.07–0.70).
INB alone was not tested in Experiment 3. Although the possibility remains that INB alone may have led to changes in movement kinematics similar to those observed for MP+INB, we felt that this was unlikely because INB alone did not result in significant changes in motor cortex excitability in Experiment 1.
Discussion
The principal result of the present experiments is that ischaemic limb deafferentation/deefferentation enhanced practice-dependent plasticity of the human motor cortex, whereas pretreatment with the GABAA receptor agonist lorazepam depressed it.
Site and nature of practice-dependent plasticity
MT was not changed by motor practice. MT reflects mainly membrane-related excitability of corticocortical axons (Ziemann et al., 1996b). Accordingly, MT is significantly elevated by sodium- and calcium-channel-blocking drugs (Mavroudakis et al., 1994; Ziemann et al., 1996b; Chen et al., 1997) but not by drugs interacting with the main neurotransmitters in the neocortex, GABA and glutamate (Ziemann et al., 1996a, b, 1998a; Liepert et al., 1997). Therefore, the lack of effect of motor practice on MT indicates that the nature of practice-dependent plasticity cannot be explained by an increase (non-specific) in membrane-related excitability of cortical neural elements, as has been reported in motor cortex neurones for some other forms of motor learning (Woody et al., 1991).
The increase in MEP size induced by MP+INB does not provide definite evidence for the site and nature of practice-dependent plasticity because MEP size assesses the excitability of the corticospinal system as a whole, including the corticomotor neurone and the spinal motor neurone (Devanne et al., 1997) and, therefore, cortical, subcortical and spinal mechanisms may contribute to an increase in MEP size. In contrast, PPE reflects the synaptic excitability of inhibitory and excitatory neural circuits specifically at the level of the motor cortex, and these circuits in turn control the excitability of the corticomotor neurones (Kujirai et al., 1993; Ziemann et al., 1996c). Accordingly, GABAA receptor agonists and N-methyl-d-aspartate (NMDA) receptor antagonists result in an increase in paired-pulse inhibition and a decrease in paired-pulse facilitation (Ziemann et al., 1996a, b, 1998a; Liepert et al., 1997; Schwenkreis et al., 1999; Di Lazzaro et al., 2000). A significant increase in PPE was induced by MP+INB only but not by any of the other interventions. As discussed above, this points specifically to a cortical site of this form of practice-dependent plasticity, which may be interpreted best as a shift in the balance of the synaptic efficacy of horizontal motor cortical circuits towards less inhibition and more facilitation (Kujirai et al., 1993; Ziemann et al., 1996c).
The results of Experiment 1 show that voluntary activation of the biceps muscle was necessary for practice-dependent plasticity to occur because the proprioceptive feedback from muscle contraction and elbow joint movement, when mimicked by electrical stimulation of the biceps muscle (ES+INB) or passive elbow movements (PAS+INB) without motor practice did not result in plastic changes (Fig. 1). This indicates that sensory feedback was not relevant for this particular form of practice-dependent plasticity. The main reason may be the nature of the practised ballistic elbow flexion movements, which are largely centrally preprogrammed movements (Hallett et al., 1975) that can be performed normally even in patients with severe deafferenting neuropathy (Hallett et al., 1975; Rothwell et al., 1982). Furthermore, motor cortex plasticity can occur during mental practice in the absence of actual movement and apparent sensory feedback (Pascual-Leone et al., 1995a). There is now substantial evidence that motor imagery activates the primary motor cortex similarly to real motor performance (Stephan et al., 1995; Porro et al., 1996; Abbruzzese et al., 1999) while not affecting the excitability of spinal motor neurones (Kasai et al., 1997; Hashimoto and Rothwell, 1999). This corroborates further the key role of voluntary (mental or actual) motor cortex activation in practice-dependent plasticity. This view does not bear on the well-established knowledge that other forms of motor cortex plasticity require defined sensory input for their occurrence (Hamdy et al., 1998; Ridding et al., 2000; Stefan et al., 2000).
The effects produced by MP+INB were relatively short-lived (~20 min) and may have arisen from short-term potentiation (STP)-like mechanisms. Like LTP, STP reflects activity-dependent synaptic strengthening, which depends on the activation of NMDA receptors (Anwyl et al., 1989; Castro-Alamancos and Connors, 1996). Although the role of NMDA receptor activation was not tested directly in the present experiments, one previous study showed that a similar enhancement of motor cortical output to the biceps muscle induced by rTMS of the motor cortex during INB-induced disinhibition depended on the activation of NMDA receptors because it could be blocked if the subjects were pretreated with an NMDA receptor antagonist (Ziemann et al., 1998c). Similarly, in a different experimental setting, which required the simultaneous contraction of a hand and a proximal arm muscle, practice-dependent plasticity in the form of a medial movement of the TMS-mapped hand representation (Liepert et al., 1999) could be prevented if subjects were pretreated with an NMDA receptor antagonist (Tegenthoff et al., 1999). Finally, a practice-dependent shift in the direction of TMS-evoked thumb movements (Classen et al., 1998) could also be suppressed by pretreatment with an NMDA receptor antagonist (Bütefisch et al., 2000).
In summary, it is most likely that the increase in motor cortex excitability of the biceps representation induced by MP+INB in the present study, in particular the increase in PPE, reflects STP-like synaptic plasticity. Other investigators have similarly proposed that synaptic cortical plasticity underlies motor learning (Donoghue et al., 1996; Asanuma and Pavlides, 1997; Rioult-Pedotti et al., 1998).
Mechanisms of the INB-induced enhancement of practice-dependent plasticity
Multimetabolite magnetic resonance spectroscopy has provided evidence that limb deafferentation/deefferentation leads to a rapid decrease in GABA content in the sensorimotor cortex contralateral to INB (Levy et al., 1999). The decrease in GABA became significant 0–10 min before complete ischaemic motor nerve block had been achieved, i.e. ~30 min into the deafferentation procedure. This is matched by the time course of changes in biceps MEP amplitudes, which also start to increase at around the time of completion of ischaemic motor nerve block (Ridding and Rothwell, 1997). The possibility of very rapid deefferentation-induced changes in cortical inhibition is supported by experiments in rats, which showed that transection of the facial nerve resulted, within 10 min, in disinhibition of the deefferented motor cortex when the animals were tested with paired intracortical microstimulation (Farkas et al., 2000).
The mechanism of the rapid decrease in GABA level is unknown. GABA is produced in the nerve terminals of GABAergic neurones from glutamate and glutamic acid by glutamic acid decarboxylase and is catabolized by GABA transaminase (GABA-T) (Tillakaratne et al., 1995). Magnetic resonance spectroscopy data have shown that the level of glutamate does not change significantly during ischaemic deafferentation (Levy et al., 1999). Therefore, downregulation of glutamic acid decarboxylase is unlikely to account for the decrease in GABA. Another magnetic resonance spectroscopy study demonstrated that GABA increased in the human cortex by >40% within 2 h of administration of a single oral dose of vigabatrin (50 mg/kg), an irreversible inhibitor of GABA-T (Petroff et al., 1996). This suggests that rapid modulation of GABA-T activity provides a candidate mechanism to explain the change in GABA concentration that occurs within 1 min. In our experiments, one would then propose a deafferentation-induced increase in GABA-T activity to explain the rapid decrease in GABA level.
Synaptic plasticity in the motor cortex depends strongly on the activity of GABA-related inhibition. Experiments on slices of rat motor cortex showed that the successful induction of LTP, in this case by repetitive electrical microstimulation, required a reduction in local cortical inhibition by iontophoretic application of the GABAA receptor antagonist bicuculline (Hess and Donoghue, 1994; Hess et al., 1996). As in the present experiments, the effects of rTMS on motor cortex excitability were also dramatically enhanced in the presence of INB-induced cortical disinhibition (Ziemann et al., 1998b).
Conversely, increasing the activity of GABA-related cortical inhibition by pretreatment with a GABAA receptor agonist resulted in a significant reduction in practice-dependent plasticity in the thumb-movement paradigm (Bütefisch et al., 2000). The present experiments extend this by demonstrating a switch from an increase to a decrease in PPE as a consequence of pretreatment with the GABAA receptor agonist lorazepam (Fig. 2B). This suggests that, beyond a mere suppression of STP-like plasticity, cortical synapses can be modified bidirectionally. A similar phenomenon was observed in slices of rat neocortex, in which LTP switched to LTD as a function of decreasing postsynaptic excitability (Artola et al., 1990; Bear and Kirkwood, 1996). Furthermore, LTD was specifically induced by afferent stimulation in the rat visual cortex if the GABAA receptor agonist muscimol was added (Kato and Yoshimura, 1993). Finally, in some subjects practising simultaneous contractions of a hand muscle and an upper arm muscle there was a lateral shift of the TMS-mapped hand representation after pretreatment with lorazepam (Tegenthoff et al., 1999). This is opposite to the medial shift observed under drug-free conditions (Liepert et al., 1999; Tegenthoff et al., 1999). The present experiments supplement this finding of opposite effects of motor practice as a function of the amount of GABA-related inhibition by suggesting a switch between LTP- (or STP-) and LTD-like synaptic plasticity as the mechanism responsible.
In summary, the present findings from Experiments 1 and 2 provide evidence for an important role of GABA-related cortical inhibition in bidirectionally modulating practice-dependent plasticity in the human motor cortex. The findings support the current view, which is based mainly on animal experiments, that GABA is key in the mechanisms and modulation of plasticity in the adult mammalian central nervous system (Jacobs and Donoghue, 1991; Jones, 1993).
Behavioural relevance of practice-dependent plasticity and its modulation
The results of Experiment 3 show that the increase in TMS-evoked motor cortical output in the MP+INB condition was not merely an epiphenomenon but was associated with a parallel improvement in behavioural measures (Fig. 3).
The extent to which one can make use of the GABA-related modulation of behaviourally relevant practice-dependent plasticity in the clinical situation is a crucial question. We have recently conducted a set of experiments in chronic stroke patients in order to facilitate the rehabilitation of hand function (W. Muellbacher, C. Richards, U. Ziemann, G. Wittenberg, D. Weltz, B. Boroojerdi, L. G. Cohen and M. Hallett, unpublished). These patients were asked to practise a pincer grip between the thumb and index finger of the paretic hand. After an initial mild improvement in maximum grip force and peak acceleration between the two fingers, motor performance quickly reached a plateau. The patients then underwent temporary anaesthetic block of the upper brachial plexus on the paretic side, resulting in selective deafferentation/deefferentation of the upper arm. This was done, as in the present experiments, in order to disinhibit the affected contralateral motor cortex; however, in the present experiments disinhibition was achieved by deafferentation/deefferentation of the hand. During this intervention, the patients resumed practice of the pincer grip. This resulted in a dramatic further increase in the MEP size of the thumb flexor muscle, maximum grip force and peak acceleration above the previously reached plateau level. In contrast, an increase in cortical inhibition is probably detrimental for the recovery of function, as shown in animal experiments (Hernandez and Schallert, 1988; Hernandez et al., 1989) and by anecdotal experience in stroke patients (for review, see Goldstein, 1998).
In conclusion, our findings suggest that GABA-related cortical inhibition can be manipulated to modulate plasticity of the human cortex. This leads to the prediction that practice-dependent cortical plasticity and associated changes in behaviour, such as perceptual and motor learning and the recovery of function after lesions, can be enhanced when concomitant measures are taken to reduce GABA-related inhibitory mechanisms in the cortex.
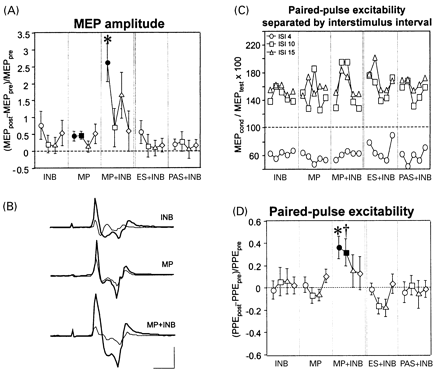
Changes in TMS-evoked motor cortical output to the biceps muscle as induced by different interventions. (A) MEP amplitude at the end of intervention (circles) and 20 min (squares), 40 min (triangles), and 60 min (diamonds) later are given as increments of the preintervention measurements (mean ± standard error). Filled symbols indicate significant differences from zero (P < 0.05). *Different from all other interventions at this time point (P < 0.05). (B) EMG recordings (averages of 10 trials) of the biceps MEP of one subject before (thin lines) and at the end of intervention (thick lines). Calibration bars, 15 ms (horizontal) and 0.25 mV (vertical). (C) Intervention-induced changes in paired-pulse excitability (PPE) shown separately for the three interstimulus intervals of 4 ms (circles), 10 ms (squares) and 15 ms (triangles). The five data points for each interval and intervention refer to the time points before intervention, late into intervention and 20, 40 and 60 min after the end of intervention. (D) Intervention-induced changes in PPE. †Different from all interventions except INB (P < 0.05). Other conventions as in panel A. INB = ischaemic nerve block at the forearm; MP = motor practice; MP+INB, ES+INB and PAS+INB = motor practice, electrical stimulation of the biceps muscle and passive elbow flexion movements, respectively, during INB.
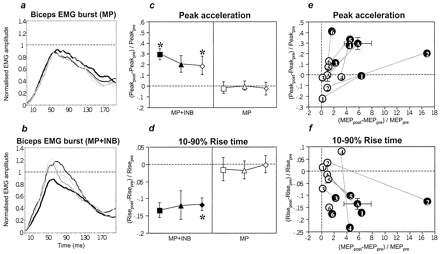
(A and B) Grand averages across all subjects (n = 6) of the individually normalized (EMG peak amplitude before intervention set to 1.0) EMG burst of the biceps muscle during maximal ballistic elbow flexion movement (A, MP alone; B, MP+INB). The thick lines show EMG data before intervention; the thin black thick and medium lines show data obtained 10, 30 and 50 min after the end of intervention, respectively. (C and D) Changes in kinematics (peak acceleration, 10–90% rise time) of maximum ballistic elbow flexion movements induced by motor practice without (MP) and with (MP+INB) ischaemic hand deafferentation. Post-measurements 10 min (squares), 30 min (triangles) and 50 min (diamonds) after the end of intervention are shown as increments of the pre-measurement (mean ± standard error). Other conventions as in Fig. 1A and B. (E and F) Individual increments in peak acceleration and rise time (10 min after intervention) are plotted against increments in biceps MEP amplitude (at the end of intervention). Each circle denotes one subject (1–6) and the average (A, ± standard error) measured in the MP (white circles) and MP+INB experiment (black circles).
This work was supported by grant Zi 542/1-1 from the Deutsche Forschungsgemeinschaft awarded to U.Z. We thank M. Rogawski and S. P. Wise for helpful discussion.
References
Abbruzzese G, Assini A, Buccolieri A, Marchese R, Trompetto C. Changes of intracortical inhibition during motor imagery in human subjects.
Anwyl R, Mulkeen D, Rowan MJ. The role of N-methyl-D-aspartate receptors in the generation of short-term potentiation in the rat hippocampus.
Artola A, Brocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex.
Asanuma H, Pavlides C. Neurobiological basis of motor learning in mammals. [Review].
Bear MF, Kirkwood A. Bidirectional plasticity of cortical synapses. In: Fazeli S, Collingridge GL, editors. Cortical plasticity. LTP and LTD. Oxford: BIOS Scientific; 1996. p. 191–205.
Boroojerdi B, Cohen LG, Petroff OA, Rothman DL. Mechanisms of light deprivation-induced enhancement of visual cortex excitability [abstract].
Borsook D, Becerra L, Fishman S, Edwards A, Jennings CL, Stojanovic M, et al. Acute plasticity in the human somatosensory cortex following amputation.
Brasil-Neto JP, Cohen LG, Pascual-Leone A, Jabir FK, Wall RT, Hallett M. Rapid reversible modulation of human motor outputs after transient deafferentation of the forearm: a study with transcranial magnetic stimulation.
Brasil-Neto JP, Valls-Sole J, Pascual-Leone A, Cammarota A, Amassian VE, Cracco R, et al. Rapid modulation of human cortical motor outputs following ischaemic nerve block.
Bütefisch CM, Davis BC, Wise SP, Sawaki L, Kopylev L, Classen J, et al. Mechanisms of use-dependent plasticity in the human motor cortex.
Castro-Alamancos MA, Connors BW. Short-term synaptic enhancement and long-term potentiation in neocortex.
Chen R, Samii A, Canos M, Wassermann EM, Hallett M. Effects of phenytoin on cortical excitability in humans.
Classen J, Liepert J, Wise SP, Hallett M, Cohen LG. Rapid plasticity of human cortical movement representation induced by practice.
Cohen LG, Ziemann U, Chen R, Classen J, Hallett M, Gerloff C, et al. Studies of neuroplasticity with transcranial magnetic stimulation. [Review].
Devanne H, Lavoie BA, Capaday C. Input-output properties and gain changes in the human corticospinal pathway.
Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A, et al. Magnetic transcranial stimulation at intensities below active motor threshold activates intracortical inhibitory circuits.
Di Lazzaro V, Oliviero A, Meglio M, Cioni B, Tamburrini G, Tonali P, et al. Direct demonstration of the effect of lorazepam on the excitability of the human motor cortex.
Donoghue JP, Suner S, Sanes JN. Dynamic organization of primary motor cortex output to target muscles in adult rats. II. Rapid reorganization following motor nerve lesions.
Donoghue JP, Leibovic S, Sanes JN. Organization of the forelimb area in squirrel monkey motor cortex: representation of digit, wrist, and elbow muscles.
Donoghue JP, Hess G, Sanes JN. Substrates and mechanisms for learning in motor cortex. In: Bloedel JR, Ebner TJ, Wise SP, editors. Acquisition of motor behavior in vertebrates. Cambridge (MA): MIT Press; 1996. p. 363–86.
Farkas T, Perge J, Kis Z, Wolff JR, Toldi J. Facial nerve injury-induced disinhibition in the primary motor cortices of both hemispheres.
Fromm C, Evarts EV. Relation of size and activity of motor cortex pyramidal tract neurons during skilled movements in the monkey.
Hallett M, Shahani BT, Young RR. EMG analysis of stereotyped voluntary movements in man.
Hamdy S, Rothwell JC, Aziz Q, Singh KD, Thompson DG. Long-term reorganization of human motor cortex driven by short-term sensory stimulation.
Hashimoto R, Rothwell JC. Dynamic changes in corticospinal excitability during motor imagery.
Hernandez TD, Schallert T. Seizures and recovery from experimental brain damage.
Hernandez TD, Jones GH, Schallert T. Co-administration of Ro 15-1788 prevents diazepam-induced retardation of recovery of function.
Hess G, Donoghue JP. Long-term potentiation of horizontal connections provides a mechanism to reorganize cortical motor maps.
Hess G, Donoghue JP. Long-term depression of horizontal connections in rat motor cortex.
Hess G, Donoghue JP. Long-term potentiation and long-term depression of horizontal connections in rat motor cortex. [Review].
Hess G, Aizenman CD, Donoghue JP. Conditions for the induction of long-term potentiation in layer II/III horizontal connections of the rat motor cortex.
Huntley GW. Correlation between patterns of horizontal connectivity and the extent of short-term representational plasticity in rat motor cortex.
Huntley GW, Jones EG. Relationship of intrinsic connections to forelimb movement representations in monkey motor cortex: a correlative anatomic and physiological study.
Jacobs KM, Donoghue JP. Reshaping the cortical motor map by unmasking latent intracortical connections.
Jones EG. GABAergic neurons and their role in cortical plasticity in primates. [Review].
Kaas JH. Plasticity of sensory and motor maps in adult mammals. [Review].
Kaas JH, Merzenich MM, Killackey HP. The reorganization of somatosensory cortex following peripheral nerve damage in adult and developing mammals. [Review].
Kasai T, Kawai S, Kawanishi M, Yahagi S. Evidence for facilitation of motor evoked potentials (MEPs) induced by motor imagery.
Kato N, Yoshimura H. Tetanization during GABAA receptor activation induces long-term depression in visual cortex slices.
Kolarik RC, Rasey SK, Wall JT. The consistency, extent, and locations of early-onset changes in cortical nerve dominance aggregates following injury of nerves to primate hands.
Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, et al. Corticocortical inhibition in human motor cortex.
Levy LM, Ziemann U, Chen R, Cohen LG. Rapid modulation of GABA in human cortical plasticity demonstrated by magnetic resonance spectroscopy [abstract].
Liepert J, Schwenkreis P, Tegenthoff M, Malin J-P. The glutamate antagonist riluzole suppresses intracortical facilitation.
Liepert J, Terborg C, Weiller C. Motor plasticity induced by synchronized thumb and foot movements.
Mavroudakis N, Caroyer JM, Brunko E, Zegers de Beyl D. Effects of diphenylhydantoin on motor potentials evoked with magnetic stimulation.
Merzenich MM, Kaas JH. Reorganization of mammalian somatosensory cortex following peripheral nerve injury.
Merzenich MM, Kaas JH, Wall JT, Sur M, Nelson RJ, Felleman DJ. Progression of change following median nerve section in the cortical representation of the hand in areas 3b and 1 in adult owl and squirrel monkeys.
Mills KR, Kimiskidis V. Motor cortex excitability during ballistic forearm and finger movements.
Nudo RJ, Milliken GW, Jenkins WM, Merzenich MM. Use-dependent alterations of movement representations in primary motor cortex of adult squirrel monkeys.
Nudo RJ, Wise BM, SiFuentes F, Milliken GW. Neural substrates for the effects of rehabilitative training on motor recovery after ischemic infarct.
Pascual-Leone A, Cammarota A, Wassermann EM, Brasil-Neto JP, Cohen LG, Hallett M. Modulation of motor cortical outputs to the reading hand of Braille readers.
Pascual-Leone A, Grafman J, Hallett M. Modulation of cortical motor output maps during development of implicit and explicit knowledge.
Pascual-Leone A, Nguyet D, Cohen LG, Brasil-Neto JP, Cammarota A, Hallett M. Modulation of muscle responses evoked by transcranial magnetic stimulation during the acquisition of new fine motor skills.
Pascual-Leone A, Wassermann EM, Sadato N, Hallett M. The role of reading activity on the modulation of motor cortical outputs to the reading hand in Braille readers.
Petroff OA, Rothman DL, Behar KL, Collins TL, Mattson RH. Human brain GABA levels rise rapidly after initiation of vigabatrin therapy.
Porro CA, Francescato MP, Cettolo V, Diamond ME, Baraldi P, Zuiani C, et al. Primary motor and sensory cortex activation during motor performance and motor imagery: a functional magnetic resonance imaging study.
Rao SM, Binder JR, Hammeke TA, Bandettini PA, Bobholz JA, Frost JA, et al. Somatotopic mapping of the human primary motor cortex with functional magnetic resonance imaging.
Ridding MC, Brouwer B, Miles TS, Pitcher JB, Thompson PD. Changes in muscle responses to stimulation of the motor cortex induced by peripheral nerve stimulation in human subjects.
Ridding MC, Rothwell JC. Reorganisation in human motor cortex.
Ridding MC, Rothwell JC. Stimulus/response curves as a method of measuring motor cortical excitability in man.
Rioult-Pedotti M-S, Friedman D, Hess G, Donoghue JP. Strengthening of horizontal cortical connections following skill learning.
Rossini PM, Barker AT, Berardelli A, Caramia MD, Caruso G, Cracco RQ, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord and roots: basic principles and procedures for routine clinical application. Report of an IFCN committee. [Review].
Rothwell JC, Traub MM, Day BL, Obeso JA, Thomas PK, Marsden CD. Manual motor performance in a deafferented man.
Sanes JN, Donoghue JP. Static and dynamic organization of motor cortex. [Review].
Sanes JN, Donoghue JP. Plasticity and primary motor cortex. [Review].
Sanes JN, Suner S, Lando JF, Donoghue JP. Rapid reorganization of adult rat motor cortex somatic representation patterns after motor nerve injury.
Sanes JN, Donoghue JP, Thangaraj V, Edelman RR, Warach S. Shared neural substrates controlling hand movements in human motor cortex.
Schwenkreis P, Witscher K, Janssen F, Addo A, Dertwinkel R, Zenz M, et al. Influence of the N-methyl-D-aspartate antagonist memantine on human motor cortex excitability.
Silva AC, Rasey SK, Wu X, Wall JT. Initial cortical reactions to injury of the median and radial nerves to the hands of adult primates.
Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation.
Stephan KM, Fink GR, Passingham RE, Silbersweig D, Ceballos-Baumann AO, Frith CD, et al. Functional anatomy of the mental representation of upper extremity movements in healthy subjects.
Tegenthoff M, Witscher K, Schwenkreis P, Liepert J. Pharmacological modulation of training-induced plastic changes in human motor cortex.
Tillakaratne NJ, Medina-Kauwe L, Gibson KM. Gamma-aminobutyric acid (GABA) metabolism in mammalian neural and nonneural tissues. [Review].
Tinazzi M, Zanette G, Polo A, Volpato D, Manganotti P, Bonato C, et al. Transient deafferentation in humans induces rapid modulation of primary sensory cortex not associated with subcortical changes: a somatosensory evoked potential study.
Wassermann EM, McShane LM, Hallett M, Cohen LG. Noninvasive mapping of muscle representations in human motor cortex.
Weinberger NM. Dynamic regulation of receptive fields and maps in the adult sensory cortex. [Review].
Woody CD, Gruen E, Birt D. Changes in membrane currents during Pavlovian conditioning of single cortical neurons.
Ziemann U, Lönnecker S, Steinhoff BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man.
Ziemann U, Lönnecker S, Steinhoff BJ, Paulus W. Effects of antiepileptic drugs on motor cortex excitability in humans: a transcranial magnetic stimulation study.
Ziemann U, Rothwell JC, Ridding MC. Interaction between intracortical inhibition and facilitation in human motor cortex.
Ziemann U, Chen R, Cohen LG, Hallett M. Dextromethorphan decreases the excitability of the human motor cortex.
Ziemann U, Corwell B, Cohen LG. Modulation of plasticity in human motor cortex after forearm ischemic nerve block.


{kind=link}
{kind=link}
{kind=link}