




Abstract
Mutations in CuZn-superoxide dismutase (CuZn-SOD) have been linked to familial amyotrophic lateral sclerosis (ALS), and motor neurone death is caused by the gain of a toxic property of the mutant protein. Here we determined amounts, activity and molecular forms of CuZn-SOD in CSF from ALS patients carrying the D90A and other CuZn-SOD mutations and patients without such mutations. There were no differences in amount of protein and enzymic activities of CuZn-SOD between 37 neurological controls, 54 sporadic and 12 familial ALS cases, and 10 cases homozygous for the D90A mutation. Three cases heterozygous for the A89V, S105L and G114A CuZn-SOD mutations showed low amounts of CuZn-SOD. There was no evidence for accumulation of inactive protein in any of the groups. Immunoblots showed no evidence for the presence of any precipitates or other molecular forms of CuZn-SOD with higher molecular weight in the groups. About 25% of the CuZn-SOD subunits in CSF from controls shows an N-terminal truncation. This truncated portion does not differ between controls and ALS groups not carrying CuZn-SOD mutations, but is 70% larger in samples from D90A homozygous ALS patients. The findings suggest an essentially normal amount and activity of D90A mutant CuZn-SOD in CNS tissues of ALS cases. The increased occurrence of N-terminally truncated mutant subunits may indicate a difference in degradation routes compared with the wild-type enzyme, resistance against subsequent proteolytic steps and/or a compromised downstream proteolytic machinery. Molecular fragments accumulated to a greater extent from the D90A mutant enzyme might contribute to the motor neurone degeneration. We also determined the other SOD isoenzymes: in the controls, CuZn-SOD contributed 75%, extracellular SOD 25% and Mn-SOD <5% of the total SOD activity. There was no difference in the amount of extracellular SOD between any of the groups.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative syndrome characterized by adult-onset progressive loss of motor neurones in the motor cortex, brainstem and medulla spinalis causing muscular wasting, paresis and inevitable death. About 10% of ALS cases are familial (FALS) (Haverkamp et al., 1995) and in ~20% of these the disease has been associated with mutations in the CuZn-superoxide dismutase (SOD, EC 1.15.1.1) gene (Andersen et al., 2000). About 90 different mutations have been identified, with all but one showing a dominant pattern of heredity, and in most cases a significant reduction in CuZn-SOD activity in erythrocytes (Deng et al., 1993), other cell types (Tsuda et al., 1994) and CNS tissue (Bowling et al., 1995). The D90A CuZn-SOD mutation shows some deviant features. Some 80 cases, apparently sporadic or members of some 20 different families, homozygous for the mutation have been found up to now (Andersen et al., 1996; Boukaftane et al., 1998). The disease in these recessive cases shows a milder phenotype (Andersen et al., 1996), and the CuZn-SOD activity is essentially normal in erythrocytes. The mechanism by which the CuZn-SOD mutations cause motor neurone degeneration is not understood, but the collective evidence (Gurney et al., 1994; Andersen et al., 1995; Reaume et al., 1996) suggests a toxic gain of function rather than loss of function.
In some other neurodegenerative diseases, such as Alzheimer's disease (Goate et al., 1991), Parkinson's disease (Polymeropoulus et al., 1997) and Creutzfeld–Jacob's disease (Prusiner et al., 1998), the protein mutated in inherited cases is also involved in the disease in sporadic cases. Furthermore, alterations in the molecular forms of the proteins can be demonstrated, and these seem to be involved in the disease processes. A similar situation may pertain to ALS. A way to mirror the CNS is to analyse the CSF. In the present study, we determined the amounts, activities and molecular forms of CuZn-SOD in CSF from 10 homozygous ALS cases, one case heterozygous for the D90A mutation, three cases heterozygous for other mutations, and 12 FALS and 54 sporadic ALS (SALS) cases not carrying such mutations, and compared them with 37 neurological controls.
Material and methods
Patients and controls
CSF was collected from 54 SALS and 12 FALS cases without CuZn-SOD mutations, 10 homozygous ALS cases, one ALS case heterozygous for the D90A CuZn-SOD mutation, and one case each heterozygous for the A89V, S105L and G114A mutations. Thirty-seven cases with headache or other neurological symptoms for which no underlying neurological disease could be diagnosed served as controls. All ALS cases were genotyped for CuZn-SOD mutations as described (Andersen et al., 1997). After tapping, the samples were immediately frozen and kept at –80°C. The patients' consent was obtained according to the declaration of Helsinki and the study was approved by the medical research ethics committee at Umeå University.
SOD analysis
The SOD activity was determined with the potassium superoxide assay, and 3 mM cyanide was used for the distinction between the sensitive CuZn-SOD and extracellular SOD (EC-SOD), and the resistant manganese-SOD (Mn-SOD) (Marklund, 1976, 1985). CuZn-SOD protein and EC-SOD protein were determined with enzyme-linked immunosorbent assay (ELISA). The activities of these isoenzymes were calculated from their specific activities, 4.4 ng/U for CuZn-SOD and 8.6 ng/U for EC-SOD. The D90A mutant CuZn-SOD shows a lower reactivity in the ELISA, 2.9 ng/U (Andersen et al., 1998).
The CSF protein content was determined with a Coomassie brilliant blue G-250 assay, standardized with human serum albumin.
Immunoblotting
The CSF samples were subjected to SDS–PAGE (sodium dodecylsulphate–polyacrylamide gel electrophoresis, 12% polyacrylamide; Bio-Rad, Hercules, Calif., USA) and electroblotted onto polyvinylidine difluoride membranes (Amersham Pharmacia Biotech, Uppsala, Sweden). Blots were probed with antibodies and chemiluminescence was generated using Supersignal West Dura substrate (Pierce, Rockford, Ill., USA). Bands were visualized on film or by using a Fluor-S Multiimager and Quantity One software (Bio-Rad, Hercules, Calif., USA). The primary antibodies used were polyclonal rabbit antibodies raised against keyhole limpet haemocyanin-coupled peptides corresponding to amino acids 3–20, 24–39, 43–57, 58–72, 80–96, 100–115 and 131–153 in the human CuZn-SOD sequence. The rabbit antisera were purified on protein A–Sepharose (Amersham Pharmacia Biotech, Uppsala, Sweden) followed by purification on a Sulfolink coupling gel (Pierce, Rockford, Ill., USA) with the corresponding coupled peptides. Secondary antibodies were horseradish peroxidase-labelled anti-rabbit IgG antibody or biotinylated anti-rabbit IgG antibody and horseradish peroxidase-labelled streptavidin (Amersham Pharmacia Biotech, Uppsala, Sweden).
Results
Activities and amounts of SOD isoenzymes
The results of the quantitative SOD isoenzyme activity and protein analyses are summarized in Table 1. In the controls, almost all the SOD activity was cyanide sensitive, i.e. produced by CuZn-SOD and EC-SOD. Using the specific activities for CuZn-SOD and EC-SOD, it can be calculated from the ELISA data that the former accounts for 73% of the SOD activity and the latter 23%. Regarding the low cyanide-resistant SOD activity, which is produced by the mitochondrial Mn-SOD, it should be noted that the SOD assay is 10-fold less sensitive for that activity than for the cyanide-sensitive isoenzymes (Marklund, 1976, 1985). Mn-SOD may account for up to 5% of the SOD activity of control CSF.
The amounts of CuZn-SOD were greater in the SALS and FALS cases without CuZn-SOD mutations than in the controls and the D90A homozygous FALS group. These differences appear to be explained by the greater ages in the SALS and FALS groups and the increases with age in the amount of CuZn-SOD in the CSF seen in these groups (r2 = 0.18, P < 0.01 and r2 = 0.27, P < 0.09, respectively) (Fig. 1). It is apparent from this figure that there is no difference in the amount of mutant CuZn-SOD in the D90A homozygous FALS group and of wild-type enzyme in the ALS cases not carrying CuZn-SOD mutations. The amounts of CuZn-SOD in the A89V, S105L and G114A heterozygotes were roughly half those seen in other ALS cases (Table 1). The single D90A heterozygote also showed a low activity.
There was no difference in the amount of EC-SOD between the three groups. As a check for the internal consistency between the SOD activity and SOD protein assays, the combined calculated activities of CuZn-SOD and EC-SOD were divided by the total measured cyanide-sensitive activities. These ratios were close to unity in all groups (Table 1). This finding also indicates that there were no significant accumulations of inactive SOD protein in any of the groups, at least as could be detected by the ELISAs.
Analysis by immunoblotting
CSF from all cases carrying CuZn-SOD mutations was analysed by immunoblotting and compared with nine controls, nine SALS and nine FALS cases, using two different antipeptide antibodies directed against amino acids 43–57 and 58–72. There was no evidence for the presence of aggregates in the stacking gel, or of smearing or other evidence for high-molecular weight materials in the separation part of the gels. As previously demonstrated (Marklund et al., 1997), the D90A mutant showed a higher mobility than the wild-type enzyme (Fig. 2A). The major novel finding was a relatively strong band with an apparent molecular weight 2.5 kDa lower than that of the full-length subunit, which was seen in all the analysed CSF samples. A band with a similar mobility is also found regularly in CNS extracts, but is barely visible. It could be visualized with all antipeptide antibodies except that directed against amino acids 3–20 in the CuZn-SOD sequence, suggesting that it represents a subunit truncated in the N-terminal end (Fig. 2B). There was no difference in the amount of the low-molecular weight band between CSF samples that had been divided into one part snap-frozen after tapping and other parts stored for 24 h at room or refrigerator temperature, suggesting that it does not represent a storage artefact (data not shown). The relative amount of the truncated subunit did not differ between controls, SALS and FALS cases, but it was considerably larger in the D90A mutant FALS cases (Table 2). The immunoblot of the D90A heterozygote CSF sample showed apparently equal amounts of the full-length subunits, and truncated forms of both types of subunits appeared to be present. The samples from the heterozygous A89V, S105L and G114A mutant ALS cases showed lower amounts of the full-length band (cf. Table 1), and the relative amounts of the truncated bands appeared to be more similar to the ratios seen in the D90A homozygotic samples. No other differences were found in immunoblots from these samples.
Discussion
The concentration of CuZn-SOD in CSF should be positively dependent on the concentration of the enzyme in the cytosol of cells in the CNS and the rates of basal background leakage and enhanced leakage from compromised cells, and inversely dependent on the rate of turnover of CSF. There was probably no major alteration in CSF turnover or vascular permeability in the ALS groups since there was no significant alteration in the amount of CSF protein compared with controls. Taking the differences in age between the groups into account, there appear to be no differences in CuZn-SOD content between the controls, the ALS and FALS groups and the D90A homozygous FALS cases. Although there should be some increased leakage of CuZn-SOD from compromised motor neurones, the disease has a slow course and afflicts only a limited portion of the cells in the CNS. In more acute diseases such as stroke, afflicting a large portion of the CNS, several fold increases in the amount of CuZn-SOD in the CSF have been found (Strand and Marklund, 1992; Yoshida et al., 1994)
The amount of mutant CuZn-SOD in CSF from patients carrying CuZn-SOD mutations has not been analysed previously. The present findings suggest that the content and activity of D90A mutant CuZn-SOD in CNS tissue are similar to those of controls and other ALS cases, in accordance with previous findings of a high activity in erythrocytes (Andersson et al., 1998) and a high stability of the isolated mutant enzyme (Marklund et al., 1997). In addition to the common recessive D90A ALS families, several pedigrees with the D90A mutation and a dominant heredity have also been reported (reviewed in Andersen et al., 2000). These dominant cases in general seem to have a more severe phenotype than that seen in the recessive cases. Haplotype analysis of families from around the world suggests that all recessive cases share a common founder, whereas the dominant cases have several founders. This indicates that there may exist a protective genetic difference tightly linked to the CuZn-SOD locus in families with the recessive disease (Al-Chalabi et al., 1998). Findings in both man (Hayward et al., 1998) and in transgenic mice (Gurney et al., 1994) indicate that there is a gene dosage effect of the CuZn-SOD mutations on disease expression. The present results suggest that the putative linked protective factor does not act by simply downregulating the synthesis of D90A mutant CuZn-SOD in the CNS.
Regarding molecular forms of CuZn-SOD in ALS, we found no evidence for the presence of precipitates or other altered forms with larger molecular weights in any of the CSF samples. Since high-molecular weight material would be expected to be retained more strongly, the finding does not preclude the presence of such material in cells in the CNS. The major novel alteration found was the low-molecular weight band, which probably represents a subunit with an N-terminal truncation. A band with similar mobility was seen regularly in CNS extracts but was much less abundant. The reason for the high concentration in CSF is unclear, but the truncated protein may, for some reason, leak into the CSF more easily than the full-length protein. The amount did not differ between the controls and SALS and FALS groups carrying the wild-type enzyme, suggesting that it is not involved in the disease in these cases. The analysis of the A89V, S105L and G114A heterozygotes indicated lowered amounts of CuZn-SOD, but the relative amounts of the low-molecular weight portion tended to be high. In general, the CuZn-SOD activity is ~50% of that of controls in CNS from heterozygotes for ALS-linked mutations, owing to rapid degradation of the mutant protein (Bowling et al., 1995). It is likely that the material seen in the immunoblots from these cases represents mainly wild-type CuZn-SOD. The truncated band was, however, significantly more abundant in the homozygous D90A cases. The truncation may represent an initial step in one route of degradation. If so, the findings suggest that a larger proportion of D90A mutant enzyme than wild-type enzyme enters that route, and/or that the mutant enzyme is more resistant against subsequent proteolytic steps and/or a compromised downstream proteolytic machinery. One could speculate that toxic molecular fragments accumulated to a greater extent from the D90A mutant enzyme might contribute to the motor neurone degeneration.
This study also presents the first comprehensive analysis of the three SOD isoenzymes in CSF. CuZn-SOD accounts for ~75% of the SOD activity, EC-SOD for 25% and Mn-SOD for <5%. In human CNS, Mn-SOD accounts for ~30% of the total SOD activity (Marklund et al., 1986), and there is thus comparatively little leakage of this isoenzyme from its normal location in the mitochondrial matrix. The low activity in the assay used precluded reliable comparison between the groups in the present study. EC-SOD is a secreted protein that normally forms an equilibrium between the fluid phase and heparan sulphate proteoglycans on cell surfaces and in the connective tissue matrix. It is synthesized by a few cell types including fibroblasts, smooth muscle cells, macrophages and possibly glial cells (Marklund, 1990; Nicolai et al., 1998). EC-SOD did not differ significantly between the groups in the present study.
Analysis of SOD in CSF from ALS cases without CuZn-SOD mutations, ALS cases homozygous for the D90A mutation, heterozygous for other mutations and from controls
| Age (years) | Direct assay of activity | Activity calculated from ELISA | Ratio ELISA/activity | Protein (mg/ml) | |||
|---|---|---|---|---|---|---|---|
| Total SOD (U/ml) | SOD in cyanide (U/ml) | EC-SOD (U/ml) | CuZn-SOS (U/ml) | ||||
| SOD enzymic activity was determined with the potassium superoxide assay. CuZn-SOD and EC-SOD protein were determined by ELISA, and the corresponding enzymic activities were calculated using the specific activities of the isoenzymes; see Material and methods. The ratios between the sum of these activities and the measured cyanide-sensitive activities were calculated for all patients, as a check for the internal consistency of the various assays. The data are presented as means ± standard deviation. One-way ANOVA (analysis of variance) with Tukey's honestly SD post hoc test found no significant differences between any of the groups. Pair-wise comparison with the Mann–Whitney non-parametric U test found the total SOD activity of the controls to be significantly lower than that of the SALS and FALS groups, P < 0.015 and P < 0.014, respectively. The CuZn-SOD activities in the controls and D90A homozygotes were significantly lower than in the FALS group, P < 0.05 and P < 0.017, respectively. | |||||||
| Control (n=37) | 52.1±15.4 | 38.9±10.6 | 0.21±0.2 | 11.0±2.3 | 30.3±9.5 | 1.07±0.09 | 0.34±0.15 |
| SALS (n=54) | 60.8±9.8 | 45.0±13.8 | 0.18±0.13 | 11.9±3.6 | 33.5±12.7 | 1.01±0.11 | 0.36±0.19 |
| FALS (n=12) | 65.6±6.8 | 47.0±10.5 | 0.13±0.13 | 12.3±3.8 | 38.7±13.7 | 1.08±0.08 | 0.36±0.09 |
| D90A (n=10) | 50.8±9.8 | 39.2±8.8 | 0.17±0.15 | 11.3±2.0 | 27.3±7.4 | 0.99±0.09 | 0.40±0.11 |
| D90A | 68 | 23.5 | 0.24 | 9.42 | 18.6 | 1.19 | 0.23 |
| A89V | 57 | 32.9 | 0.57 | 12.7 | 21.8 | 1.05 | 0.21 |
| G114A | 39 | 20.6 | 0.29 | 8.6 | 14.8 | 1.14 | 0.62 |
| S105L | 55 | 23.0 | 0.39 | 10.1 | 14.6 | 1.07 | 0.41 |
| Age (years) | Direct assay of activity | Activity calculated from ELISA | Ratio ELISA/activity | Protein (mg/ml) | |||
|---|---|---|---|---|---|---|---|
| Total SOD (U/ml) | SOD in cyanide (U/ml) | EC-SOD (U/ml) | CuZn-SOS (U/ml) | ||||
| SOD enzymic activity was determined with the potassium superoxide assay. CuZn-SOD and EC-SOD protein were determined by ELISA, and the corresponding enzymic activities were calculated using the specific activities of the isoenzymes; see Material and methods. The ratios between the sum of these activities and the measured cyanide-sensitive activities were calculated for all patients, as a check for the internal consistency of the various assays. The data are presented as means ± standard deviation. One-way ANOVA (analysis of variance) with Tukey's honestly SD post hoc test found no significant differences between any of the groups. Pair-wise comparison with the Mann–Whitney non-parametric U test found the total SOD activity of the controls to be significantly lower than that of the SALS and FALS groups, P < 0.015 and P < 0.014, respectively. The CuZn-SOD activities in the controls and D90A homozygotes were significantly lower than in the FALS group, P < 0.05 and P < 0.017, respectively. | |||||||
| Control (n=37) | 52.1±15.4 | 38.9±10.6 | 0.21±0.2 | 11.0±2.3 | 30.3±9.5 | 1.07±0.09 | 0.34±0.15 |
| SALS (n=54) | 60.8±9.8 | 45.0±13.8 | 0.18±0.13 | 11.9±3.6 | 33.5±12.7 | 1.01±0.11 | 0.36±0.19 |
| FALS (n=12) | 65.6±6.8 | 47.0±10.5 | 0.13±0.13 | 12.3±3.8 | 38.7±13.7 | 1.08±0.08 | 0.36±0.09 |
| D90A (n=10) | 50.8±9.8 | 39.2±8.8 | 0.17±0.15 | 11.3±2.0 | 27.3±7.4 | 0.99±0.09 | 0.40±0.11 |
| D90A | 68 | 23.5 | 0.24 | 9.42 | 18.6 | 1.19 | 0.23 |
| A89V | 57 | 32.9 | 0.57 | 12.7 | 21.8 | 1.05 | 0.21 |
| G114A | 39 | 20.6 | 0.29 | 8.6 | 14.8 | 1.14 | 0.62 |
| S105L | 55 | 23.0 | 0.39 | 10.1 | 14.6 | 1.07 | 0.41 |
Analysis of SOD in CSF from ALS cases without CuZn-SOD mutations, ALS cases homozygous for the D90A mutation, heterozygous for other mutations and from controls
| Age (years) | Direct assay of activity | Activity calculated from ELISA | Ratio ELISA/activity | Protein (mg/ml) | |||
|---|---|---|---|---|---|---|---|
| Total SOD (U/ml) | SOD in cyanide (U/ml) | EC-SOD (U/ml) | CuZn-SOS (U/ml) | ||||
| SOD enzymic activity was determined with the potassium superoxide assay. CuZn-SOD and EC-SOD protein were determined by ELISA, and the corresponding enzymic activities were calculated using the specific activities of the isoenzymes; see Material and methods. The ratios between the sum of these activities and the measured cyanide-sensitive activities were calculated for all patients, as a check for the internal consistency of the various assays. The data are presented as means ± standard deviation. One-way ANOVA (analysis of variance) with Tukey's honestly SD post hoc test found no significant differences between any of the groups. Pair-wise comparison with the Mann–Whitney non-parametric U test found the total SOD activity of the controls to be significantly lower than that of the SALS and FALS groups, P < 0.015 and P < 0.014, respectively. The CuZn-SOD activities in the controls and D90A homozygotes were significantly lower than in the FALS group, P < 0.05 and P < 0.017, respectively. | |||||||
| Control (n=37) | 52.1±15.4 | 38.9±10.6 | 0.21±0.2 | 11.0±2.3 | 30.3±9.5 | 1.07±0.09 | 0.34±0.15 |
| SALS (n=54) | 60.8±9.8 | 45.0±13.8 | 0.18±0.13 | 11.9±3.6 | 33.5±12.7 | 1.01±0.11 | 0.36±0.19 |
| FALS (n=12) | 65.6±6.8 | 47.0±10.5 | 0.13±0.13 | 12.3±3.8 | 38.7±13.7 | 1.08±0.08 | 0.36±0.09 |
| D90A (n=10) | 50.8±9.8 | 39.2±8.8 | 0.17±0.15 | 11.3±2.0 | 27.3±7.4 | 0.99±0.09 | 0.40±0.11 |
| D90A | 68 | 23.5 | 0.24 | 9.42 | 18.6 | 1.19 | 0.23 |
| A89V | 57 | 32.9 | 0.57 | 12.7 | 21.8 | 1.05 | 0.21 |
| G114A | 39 | 20.6 | 0.29 | 8.6 | 14.8 | 1.14 | 0.62 |
| S105L | 55 | 23.0 | 0.39 | 10.1 | 14.6 | 1.07 | 0.41 |
| Age (years) | Direct assay of activity | Activity calculated from ELISA | Ratio ELISA/activity | Protein (mg/ml) | |||
|---|---|---|---|---|---|---|---|
| Total SOD (U/ml) | SOD in cyanide (U/ml) | EC-SOD (U/ml) | CuZn-SOS (U/ml) | ||||
| SOD enzymic activity was determined with the potassium superoxide assay. CuZn-SOD and EC-SOD protein were determined by ELISA, and the corresponding enzymic activities were calculated using the specific activities of the isoenzymes; see Material and methods. The ratios between the sum of these activities and the measured cyanide-sensitive activities were calculated for all patients, as a check for the internal consistency of the various assays. The data are presented as means ± standard deviation. One-way ANOVA (analysis of variance) with Tukey's honestly SD post hoc test found no significant differences between any of the groups. Pair-wise comparison with the Mann–Whitney non-parametric U test found the total SOD activity of the controls to be significantly lower than that of the SALS and FALS groups, P < 0.015 and P < 0.014, respectively. The CuZn-SOD activities in the controls and D90A homozygotes were significantly lower than in the FALS group, P < 0.05 and P < 0.017, respectively. | |||||||
| Control (n=37) | 52.1±15.4 | 38.9±10.6 | 0.21±0.2 | 11.0±2.3 | 30.3±9.5 | 1.07±0.09 | 0.34±0.15 |
| SALS (n=54) | 60.8±9.8 | 45.0±13.8 | 0.18±0.13 | 11.9±3.6 | 33.5±12.7 | 1.01±0.11 | 0.36±0.19 |
| FALS (n=12) | 65.6±6.8 | 47.0±10.5 | 0.13±0.13 | 12.3±3.8 | 38.7±13.7 | 1.08±0.08 | 0.36±0.09 |
| D90A (n=10) | 50.8±9.8 | 39.2±8.8 | 0.17±0.15 | 11.3±2.0 | 27.3±7.4 | 0.99±0.09 | 0.40±0.11 |
| D90A | 68 | 23.5 | 0.24 | 9.42 | 18.6 | 1.19 | 0.23 |
| A89V | 57 | 32.9 | 0.57 | 12.7 | 21.8 | 1.05 | 0.21 |
| G114A | 39 | 20.6 | 0.29 | 8.6 | 14.8 | 1.14 | 0.62 |
| S105L | 55 | 23.0 | 0.39 | 10.1 | 14.6 | 1.07 | 0.41 |
Ratios between the apparently N-terminally truncated and full-length subunits of CuZn-SOD in CSF
| Ratio truncated/native subunit (means ± SD) | |
|---|---|
| The ratios were determined twice for each sample by quantitative fluorimaging of the chemiluminescent immunoblots as described in Material and methods. The ratios for the D90A mutant samples were significantly greater than those of the controls (one-way ANOVA with Tukey's HSD post hoc test, P < 0.001). There were no significant differences between the FALS and SALS cases and the controls. | |
| Controls (n = 9) | 0.31 ± 0.07 |
| SALS (n = 9) | 0.26 ± 0.07 |
| FALS (n = 9) | 0.28 ± 0.08 |
| D90A (n = 10) | 0.47 ± 0.08 |
| A89V | 0.40 |
| S105L | 0.44 |
| G114A | 0.41 |
| Ratio truncated/native subunit (means ± SD) | |
|---|---|
| The ratios were determined twice for each sample by quantitative fluorimaging of the chemiluminescent immunoblots as described in Material and methods. The ratios for the D90A mutant samples were significantly greater than those of the controls (one-way ANOVA with Tukey's HSD post hoc test, P < 0.001). There were no significant differences between the FALS and SALS cases and the controls. | |
| Controls (n = 9) | 0.31 ± 0.07 |
| SALS (n = 9) | 0.26 ± 0.07 |
| FALS (n = 9) | 0.28 ± 0.08 |
| D90A (n = 10) | 0.47 ± 0.08 |
| A89V | 0.40 |
| S105L | 0.44 |
| G114A | 0.41 |
Ratios between the apparently N-terminally truncated and full-length subunits of CuZn-SOD in CSF
| Ratio truncated/native subunit (means ± SD) | |
|---|---|
| The ratios were determined twice for each sample by quantitative fluorimaging of the chemiluminescent immunoblots as described in Material and methods. The ratios for the D90A mutant samples were significantly greater than those of the controls (one-way ANOVA with Tukey's HSD post hoc test, P < 0.001). There were no significant differences between the FALS and SALS cases and the controls. | |
| Controls (n = 9) | 0.31 ± 0.07 |
| SALS (n = 9) | 0.26 ± 0.07 |
| FALS (n = 9) | 0.28 ± 0.08 |
| D90A (n = 10) | 0.47 ± 0.08 |
| A89V | 0.40 |
| S105L | 0.44 |
| G114A | 0.41 |
| Ratio truncated/native subunit (means ± SD) | |
|---|---|
| The ratios were determined twice for each sample by quantitative fluorimaging of the chemiluminescent immunoblots as described in Material and methods. The ratios for the D90A mutant samples were significantly greater than those of the controls (one-way ANOVA with Tukey's HSD post hoc test, P < 0.001). There were no significant differences between the FALS and SALS cases and the controls. | |
| Controls (n = 9) | 0.31 ± 0.07 |
| SALS (n = 9) | 0.26 ± 0.07 |
| FALS (n = 9) | 0.28 ± 0.08 |
| D90A (n = 10) | 0.47 ± 0.08 |
| A89V | 0.40 |
| S105L | 0.44 |
| G114A | 0.41 |
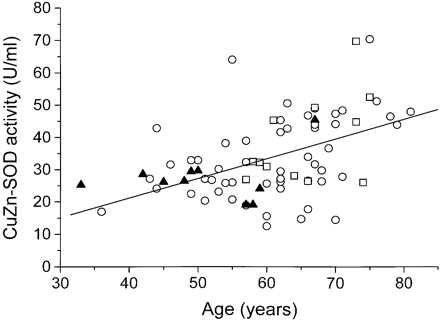
CuZn-SOD in CSF as a function of age in 10 homozygous D90A ALS cases (closed triangles), and 54 SALS cases (open circles) and 12 FALS cases (open squares) without CuZn-SOD mutations. The line shows the regression between age and CuZn-SOD for the combined 54 SALS and 12 FALS cases (r2 = 0.20, P < 0.0001).
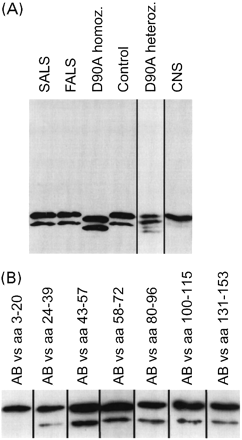
Immunoblotting of CSF. (A) CSF from one SALS, one FALS, one D90A homozyogous FALS, one D90A heterozygous ALS case and one control was immunoblotted with an antibody raised against amino acids 58–72 in the CuZn-SOD sequence. For comparison, a corresponding immunoblot from control CNS (lumbar anterior horn) is shown. (B) Immunoblots of control CSF using different rabbit antibodies raised against peptides corresponding to the indicated amino acid sequences in the CuZn-SOD subunit.
We wish to thank the patients who participated in this study, and Eva Bern, Lena Hedman, Karin Hjertkvist, Margit Lundmark, Lena Lundqvist and Agneta Öberg for expert technical assistance. The study was supported by grants from the Swedish Medical Research Council, the Swedish Medical Society, the Council of Västerbotten County, Sweden and the Swedish Association of the Neurologically Disabled.
References
Al-Chalabi A, Andersen PM, Chioza B, Shaw C, Sham PC, Robberecht W, et al. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor.
Andersen PM, Nilsson P, Ala-Hurula V, Keränen M-L, Tarvainen I, Haltia T, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase.
Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keränen M-L, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients.
Andersen PM, Nilsson P, Keränen M-L, Forsgren L, Hägglund J, Karlsborg M, et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia.
Andersen PM, Nilsson P, Forsgren L, Marklund SL. CuZn-superoxide dismutase, extracellular superoxide dismutase, and glutathione peroxidase in blood from individuals homozygous for Asp90Ala CuZn-superoxide dismutase mutation.
Andersen PM, Morita M, Brown. RH Jr. Genetics of amyotrophic lateral sclerosis: an overview. In: Brown RH Jr, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz; 2000. p. 223–50.
Boukaftane Y, Khoris J, Moulard B, Salachas F, Meininger V, Malafosse A, et al. Identification of six novel SOD1 gene mutations in familial amyotrophic lateral sclerosis.
Bowling AC, Barkowski EE, McKenna-Yasek D, Sapp P, Horvitz HR, Beal MF, et al. Superoxide dismutase concentration and activity in familial amyotrophic lateral sclerosis.
Deng H-X, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung W-Y, et al. Amyotrophic lateral sclerosis and structural defects in CuZn superoxide dismutase.
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimers disease.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human CuZn superoxide dismutase mutation.
Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population.
Hayward C, Brock DJ, Minns RA, Swingler RJ. Homozygosity of Asn86Ser mutation in the CuZn-superoxide dismutase gene produces a severe clinical phenotype in a juvenile onset case of familial amyotrophic lateral sclerosis [letter].
Marklund SL. Spectrophotometric study of spontaneous disproportionation of superoxide anion radical and sensitive direct assay for superoxide dismutase.
Marklund SL. Direct assay of superoxide dismutase with potassium superoxide. In: Greenwald RA, editor. Handbook of methods for oxygen radical research. Boca Raton (FL): CRC Press; 1998. p. 249–55.
Marklund SL. Expression of extracellular superoxide dismutase by human cell lines.
Marklund SL, Adolfsson R, Gottfries CG, Winblad B. Superoxide dismutase isoenzymes in normal brains and in brains from patients with dementia of Alzheimer type.
Marklund SL, Andersen PM, Forsgren L, Nilsson P, Ohlsson PI, Wikander G, et al. Normal binding and reactivity of copper in mutant superoxide dismutase isolated from amyotrophic lateral sclerosis patients.
Nicolai S, Willems J, Zwijsen A, Mechelen EV, Slegers H. Cyclic AMP-induced differentiation increases the synthesis of extracellular superoxide dismutase in rat C6 glioma.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease.
Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. [Review].
Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury.
Strand T, Marklund SL. Release of superoxide dismutase into cerebrospinal fluid as a marker of brain lesion in acute cerebral infarction.
Tsuda T, Munthasser S, Fraser PE, Percy ME, Rainero I, Vaula G, et al. Analysis of the functional effects of a mutation in SOD1 associated with familial amyotrophic lateral sclerosis.


{kind=link}
{kind=link}