




Abstract
Triple A (Allgrove) syndrome is characterized by achalasia, alacrima, adrenal abnormalities and a progressive neurological syndrome. Affected individuals have between two and four of these relatively common clinical problems; hence the diagnosis is often difficult in all but the classical presentation. The inheritance is autosomal recessive, and most cases of triple A have no family history. Using genetic linkage analysis in a small number of families, a locus on chromosome 12q13 was identified. The triple A gene was identified recently at this locus and called ALADIN (alacrima, achalasia, adrenal insufficiency neurologic disorder). Mutations in this gene were reported in families from North Africa and Europe. The majority of mutations were homozygous. We have identified 20 families with between two and four of the clinical features associated with the triple A syndrome. Sequencing of the triple A gene revealed five families that had a total of nine compound heterozygous mutations, and one Portuguese family (previously published) had two homozygous mutations; these changes were spread throughout the triple A gene in exons 1, 2, 7, 8, 10, 11, 12, 13 and 16, and the poly(A) tract. Those bearing mutations had the classical triple A syndrome of achalasia, alacrima, adrenal abnormalities and a progressive neurological syndrome. We identified a spectrum of associated neurological abnormalities in these cases, including pupil and cranial nerve abnormalities, frequent optic atrophy, autonomic neuropathy and upper and lower motor neurone signs including distal motor neuropathy and amyotrophy with severe selective ulnar nerve involvement. In these families, we have made genotype–phenotype correlations. Mutations in the triple A gene are thus an important cause of this clinically heterogeneous syndrome, and sequencing represents an important diagnostic investigation. Identifying further mutations and defining their phenotype along with functional protein analysis will help to characterize this neuroendocrine gene.
Introduction
The triple A (Allgrove) syndrome was first described in two pairs of siblings by Allgrove and colleagues in 1978 [Online Mendelian Inheritance in Man (OMIM) database accession number: 231550; Allgrove et al., 1978]. The disorder is characterized by achalasia, alacrima and adrenocorticotrophic hormone (ACTH)‐resistant adrenal failure. A number of associated features have been described including progressive central, peripheral and autonomic nervous system abnormalities, palmo‐plantar and punctate hyperkeratosis, short stature, osteoporosis, xerostomia, angular cheilitis, glossitis and fissured tongue, and microcephaly (Allgrove et al., 1978; Geffner et al., 1983; Stuckey et al., 1987; Dumic et al., 1991; Moore et al., 1991; Grant et al., 1992, 1993; Gazarian et al., 1995; Heinrichs et al., 1995; Hübschmann, 1995; Chu et al., 1996; Clark and Weber, 1998; Huebner et al., 1999; Zeharia et al., 1999; Bentes et al., 2001). Necropsy has been carried out in one of the pairs of original siblings published (Allgrove et al., 1978). This revealed atrophy of the adrenal cortical zona fasciculata and zona reticularis and absence of ganglion cells and nerve fibres in the lower oesophagus. No comment was made regarding the rest of the pathological examination.
A number of families have been reported in the literature. All display an autosomal recessive pattern of inheritance and most have congsanguineous parents (Allgrove et al., 1978; Geffner et al., 1983; Ehrich et al., 1987; Stuckey et al., 1987; Dumic et al., 1991; Moore et al., 1991; Grant et al., 1992, 1993; Gazarian et al., 1995; Heinrichs et al., 1995; Hübschmann, 1995; Zeharia et al., 1999; Bentes et al., 2001). Combining a number of these small families, a locus on chromosome 12q13 (Weber et al., 1996; Huebner et al., 2000) was identified. This was replicated in two other population groups (Stratakis et al., 1997; Hadj‐Rabia et al., 2000; Lee et al., 2000). Using positional cloning, the product of the triple A gene, designated ALADIN for alacrima, achalasia, adrenal insufficiency neurologic disorder, was identified recently as a WD‐repeat protein (Tullio‐Pelet et al., 2000; Handschug et al., 2001). This functionally diverse type of protein is coded for frequently in the genome consistent with triple A syndrome, which is a multi‐tissue disorder with ubiquitous gene expression; notably a particularly high expression is seen in the adrenal gland, gastrointestinal tract and brain. Mutations in this gene have also been identified in consanguineous North African and European families (Tullio‐Pelet et al., 2000; Handschug et al., 2001). Two families with compound heterozygous mutations were also identified in kindreds where the parents were unrelated (Handschug et al., 2001). An additional study of triple A families from Puerto Rico identified further families with the homozygous IVS14 + 1G→A mutation and one compound heterozygous mutation, and a Canadian family homozygous for an exon 1 point mutation (Sandrini et al., 2001).
There is significant heterogeneity in the clinical features and the types of mutation reported in families with suspected triple A syndrome. No study has looked at both the clinical manifestations and the genetic characterization in a large group of families. We therefore sequenced the triple A gene in a group of 20 families with progressive neurological problems and at least one of the triad of achalasia, alacrima and adrenal abnormalities. To look for and to characterize fully genotype–phenotype correlations in our families where mutations were identified, we re‐examined affected and unaffected individuals from these kindreds in detail carrying out repeat neurophysiology, autonomics and pupillography.
Methods
Patients
Ethics approval was obtained from the joint medical and ethics committee at The National Hospital for Neurology and Neurosurgery to carry out this genetic study. We have collected blood samples from 20 families where the proband had progressive neurological problems and at least one of the triad of achalasia, alacrima and adrenal abnormalities. Members of eight out of these 20 families had all the characteristics of triple A syndrome (see Tables 2–5 for details). A blood sample and clinical details from one of these eight families (family 6) were obtained from Portugal (Bentes et al., 2001). Individuals in the other 12 families had progressive neurological problems, consisting of predominantly motor neuropathy; 10 out of the 12 also had optic atrophy while two of 12 had retinopathy. Members of six of the 12 families had mild spasticity in the upper and lower limbs. Ten out of 12 of the families had gut abnormalities, mainly achalasia, and the other two families had alacrima. Nineteen families were patients of The National Hospital for Neurology, and one family (family 6) was from the Hospital de Santa Maria in Lisbon, Portugal (Bentes et al., 2001). All had had detailed clinical examination and investigations, including routine and specialized blood tests, thyroid function tests, CSF for cells, protein glucose and bands. MRI brain and genetic testing had been carried out to exclude the common mitochondrial mutations. Clinical neurophysiology and autonomic function tests were carried out in all families. In families with triple A mutations, further examination (by H.H. in families 1–5) of the proband and relatives was carried out. Investigations including repeat neurophysiology and pupillography were also done. All families were of European origin, the majority English; there was no history of consanguinity (Fig. 1).
Genetic sequencing
DNA was extracted from blood samples obtained from affected and unaffected individuals from families. We sequenced the entire coding region and flanking introns of the triple A gene in an affected individual from each family. The 16 exons and flanking intronic regions (see Table 1 for primer sequences) of the triple A gene were amplified by PCR. The intronic regions between exons 4 and 5, 10 and 11, 12 and 13, and 14 and 15 were small enough to allow these exons to be amplified together. PCR was carried out in a total volume of 50 µl, which contained 20 ng of DNA, 0.2 mM dNTPs, 1 U of TaqGold polymerase, 1.5 mM MgCl2, 75 mM Tris–HCl pH 9.0, 20 mM (NH4)2SO4, 0.01% Tween‐20 and 50 pmol of each primer. PCR was performed on a Perkin‐Elmer 9700 thermal cycler (Perkin Elmer, Applied Biosystems, Foster City, California, USA). The cycling consisted of denaturation at 94°C for 15 min, followed by 25 cycles of 94°C for 30 s, 60°C to 50°C touching down protocol for 30 s, and 72°C for 30 s. After that, 12 cycles of constant annealing temperature at 50°C and a final amplification at 72°C for 10 min was carried out. PCR fragments were checked on a 1% agarose gel. The PCR products were purified by using a Qiaquick purification kit (Qiagen, Hilden, Germany) and resuspended in 50 µl of deionized water. For each exon, 100 ng of amplified product was sequenced using forward and reverse primers and the BigDye Terminator cycle sequencing kit (Perkin Elmer). Sequencing was performed on an ABI377 automated sequencer; alignment and analysis were carried out with Sequence Navigator (Perkin Elmer).
Mutation analysis
DNA from parents was available in families 2, 4 and 5 and also from grandparents in family 4. Mutation analysis was carried out and the inheritance pattern was consistent with autosomal recessive with mutations segregating with disease; parents and one grandparent were clinically normal heterozygous carriers in these three compound heterozygous families (Fig. 1). Siblings were examined in families 1 (normal sister), 2 (normal brother and sister) and 4 (normal brother and sister). The affected individual in family 6 had normal parents. Mutations segregated with the disease in all families and were not present in 75 controls. One silent polymorphism was identified in the triple A gene; this change was present in controls.
Autonomic function tests and neurophysiology
Detailed autonomic screening function tests, with a particular view to determining sympathetic vasoconstrictor and cardiac parasympathetic function, were performed. These included the responses to head‐up tilt, to a series of stimuli that should raise blood pressure, to a variety of manoeuvres which would change heart rate, and to standing upright. The protocols followed were as described previously (Mathias and Bannister, 1999)
Clinical neurophysiology
Nerve conduction studies detailed in Table 5 were performed by standard methods (De Lisa et al., 1994) using a Nicolet Viking II EMG machine. Supramaximal nerve stimuli were used throughout, with a stimulus duration of 0.1 ms.
Sensory conduction in the upper limbs was recorded orthodromically with ring stimulating electrodes on the fingers and recording the evoked responses with surface electrodes over the median or ulnar nerves at the wrist. The antidromic recording technique was used in the lower limbs, stimulating the superficial peroneal and sural nerves above the ankle and recording over the nerves at the ankle.
Motor conduction was studied by recording compound muscle action potentials with surface electrodes over abductor policis brevis for the median nerve and adductor digiti minimi for the ulnar nerve, and stimulating at both the wrist and the elbow in each case. Peroneal motor conduction was measured recording from extensor digitorum brevis and stimulating the peroneal nerve at the ankle and at the fibular head.
Pupillography
This was carried out in the pupil laboratory (by S.S.) on families 1–5. Pupil diameters and their responses to light and accommodation (near) were recorded with a Whittaker/Applied Science Laboratories infrared television pupillometer as previously described (Smith et al., 1978). Pupil diameters were recorded in darkness, and near responses under dim ambient illumination. Light reflexes were induced with xenon arc or light‐emitting diode white illumination, 1 s duration at 10 s intervals, at an intensity sufficient to produce the largest possible reflex for each subject. Darkness diameters, the magnitude of light reflex responses and the rate of redilatation were compared with those obtained in healthy subjects (Smith and Smith, 1999). Pupillotonia was sought by inspection of the rate of constriction in response to continuous bright illumination.
Nerve biopsy
Nerve biopsy was only carried out in family 6, from Portugal. Sural nerve fascicular biopsies were obtained from a standard retromalleolar site. Following routine aldehyde fixation, post‐osmication, dehydration through an ascending alcohol series and resin embedding, semi‐thin sections were stained with thionin and acridine orange (Sievers, 1971).
Results
Out of the 20 families analysed, five were found to have compound heterozygous mutations and one two homozygous changes in the triple A gene. A total of 11 different mutations were found in these kindreds, of which nine were novel (Table 2). The mutations were spread throughout the triple A gene in exons 1, 2, 7, 8, 10, 11, 12, 13 and 16. The majority of mutations were insertions or deletions and created a frameshift change; two were nonsense and two were missense. A sixth family from Lisbon in Portugal (Bentes et al., 2001) had two previously unidentified homozygous mutations, one that caused the amino acid change valine to alanine at codon 313 and another that causes a deletion of ATAA in the poly(A) tract (bp 1730–1733). The exonic mutation is probably the pathogenic mutation in this family as the deletion is after the stop codon of the triple A gene, although mutations that disrupt the poly(A) tract can be pathogenic by affecting mRNA stability or splicing (Yuregir et al., 1992; Beaudoing et al., 2000). The family trees and segregation are shown in Fig. 1. Family mutation details are given in Table 2. A further sequence change was identified in exon 5 at codon 138 (GAT→GAC); this was silent and did not change an amino acid, and was also present in controls.
All individuals with mutations had progressive neurological abnormalities, achalasia, alacrima and adrenal dysfunction with varying degrees of severity. The affected individual of families 1 and 2 had less severe manifestations. The non‐neurological clinical features of affected individuals from families with triple A mutations are presented in Table 3. The clinical details of the Portuguese family (family 6) have already been published (Bentes et al., 2001). However, in brief, the affected individual had late‐onset disease starting at 25 years of age with achalasia, followed 5 years later by slowly progressive lower limb weakness. On examination, upper and motor neurone signs were detected in the upper and lower limbs; the tongue was atrophic and with fasciculations. Symptoms of autonomic dysfunction (sexual impotence, orthostatic hypotension and alacrima) appeared 11 years after the first symptoms. Adrenal impairment was recognized by laboratory testing. The mutation type in this family is different from that of the other families (Table 2). Families 3–5 presented in childhood; families 4 and 5 with hypoglycaemic seizures after diarrhoea in childhood and family 3 with alacrima. The affected individuals in families 1 and 2 presented in their teens with lower limb weakness.
Details of the neurological examination are presented in Table 4. Manifestations included bilateral optic atrophy in all families apart from family 6; the affected individuals from family 2 also had right Horner’s syndrome as well as bilateral retinopathy, and those from families 1, 3 and 6 had an atrophic tongue and spastic dysarthria. The affected individual in family 6 had a left palatal paresis, bilateral trapezius and sternocleidomastoid weakness. The two individuals from family 5 had mild mental retardation and parkinsonism; between 10 year periods of examination this had not deteriorated (Grant et al., 1992). All but one of the families (family 4) had abnormal pupils, in each instance the abnormality being bilateral and approximately symmetrical. There was pes cavus and predominantly distal weakness and wasting in the upper and lower limbs in all families. In the upper limbs, this was patchy with marked weakness in an ulnar distribution and wasting of the hypothenar eminence. The tone was generally increased in the upper and lower limbs, reflexes were brisk in the upper limbs but the ankle jerks were lost; plantar reflexes were extensor in one family and equivocal in another. Sensory examination showed loss of vibration sense only in families 1 and 2.
Neurophysiology is presented in Table 5 from the families where testing was carried out at The National Hospital for Neurology and from family 6 which was analysed in Portugal. Nerve conduction studies showed a predominant motor neuropathy in all families, predominantly axonal with mild sensory abnormalities. EMG showed chronic partial denervation. In all families analysed, there was selective involvement of ulnar nerves in the upper limbs, with the compound muscle action potential being severely affected compared with the median. Nerve conduction studies were carried out in family 5 in detail at age 10 years as well as the data given in Table 5. The sural nerve sensory action potentials were normal at age 10 years in case 5.01, but by the age of 17 years these had deteriorated and were reduced further at age 18 years. Repeat neurophysiology was carried out over between 3 and 6 years in families 1, 2 and 3; these were abnormal but there was little deterioration over this time. Conduction block and ulnar entrapment at the elbow was excluded. Central motor conduction time was prolonged in families 1, 2 and 6 but normal in family 3.
Somatosensory and visual potential examinations were also carried out in family 6 and were normal. MRI of the head and spinal cord was carried out in families 1–4 and 6 and was normal. Sural nerve biopsy was carried out in family 6, and this was normal under light microscopy.
In families 1–5, cardiovascular autonomic testing indicated that orthostatic hypotension, as defined by present criteria (20 mmHg systolic or 10 mmHg diastolic blood pressure fall), was present in families 5 and 3 (patients 5.01 and 3.02). The two were studied again at intervals of 2 and 3 years, respectively, and over this period progression was seen in relation to the degree of orthostatic hypotension, impairment of responses to pressor stimuli and parasympathetic cardiac impairment. One subject was studied at age 16 and 18 years (patient 5.01), and the other at age 33 and 39 years (patient 3.02). Patient 5.01 was on replacement hydrocortisone, and although this might have influenced the responses to some extent, it would not explain the abnormal tests of autonomic neural function. In other subjects, there was no clear evidence of autonomic dysfunction, but there were small falls in systolic blood pressure during head‐up tilt in two families, suggesting partial defects. Family 6 had cardiovascular autonomic and sudomotor testing carried out in Portugal, and these were abnormal.
Two families with the full triple A syndrome phenotype did not have mutations in the gene. Only one individual was affected in each non‐consanguineous family, but both had features of achalasia, alacrima and borderline normal adrenal problems with progressive severe neurological abnormalities. Both presented in their teenage years with lower limb weakness. The first had peripheral motor and sensory neuropathy with marked amyotrophy and cerebellar ataxia; the second had bilateral optic atrophy with distal motor neuropathy. Parents were not available for examination but were understood to have been normal.
Discussion
We collected DNA from 20 families with either a phenotype consistent with the clinical triad of triple A syndrome or a progressive neurological syndrome with one of the three features of the triple A syndrome. Eight of these 20 families had all the characteristic features of this disorder. Sequencing the triple A gene in our families revealed five families with compound heterozygous mutations and one family with homozygous mutations present (Table 2). Mutations in the triple A gene were only present in six families with a classical phenotype of achalasia, alacrima, adrenal abnormalities and a progressive neurological syndrome. These mutations are pathogenic; they segregate with the disease and they are not present in control individuals. Seven of the mutations are structurally severe frameshift or nonsense mutants and the other two cause a substitution of a conserved amino acid. In all the families with compound heterozygous mutations, one mutation was a frameshift and the other was a missense or a nonsense mutation; these previously unreported changes are predicted to cause a truncated, non‐functional protein. Two missense mutations were also identified; these two mutations have been reported previously (Handschug et al., 2001). The exon 8 (Ser263→Pro) mutation is likely to disrupt the β‐propeller structure of the gene; the exon 1 (Gln15→Lys) mutant was identified in two families and is conserved in species, segregates with the disease and may be in an area involved in ALADIN protein β‐strand stability.
Achalasia, adrenal dysfunction and alacrima were present in all six families (Table 3). Families 2 and 6 had only borderline adrenal function abnormality. There was no significant correlation between each of these manifestations. Families 1 and 2 had less severe manifestations, which is also true for their neurological features.
In our series of triple A families, we have carried out detailed neurological examination (Table 4). Cranial nerve manifestations were present in all families with varying degrees of severity. Pupil abnormalities are a common component of the triple A syndrome. At what stage these abnormalities occur is not known, but it may be pertinent that our patient with normal pupils (proband of family 4) was also the youngest by some years (17 versus 32 years or more). It is also of note that case 5.01 had normal peroneal sensory nerve action potential and compound muscle action potential at age 10 years, but this had deteriorated by age 17 years. Thus, this suggests that symptoms progress and develop in teenage and early adult years. The most frequent finding, pupillotonia, occurred in five cases. Absent, or reduced and very slow, light reflexes have already been reported in this condition (Stuckey et al., 1987; Hammami et al., 1989; Gazarian et al., 1995), observations which are compatible with our findings. Pupillotonia is considered to be a consequence of long‐term parasympathetic nerve damage (Loewenfeld and Thompson, 1967). More widespread parasympathetic abnormalities involving the cardiovascular system have been reported in an earlier follow‐up study of both patients in family 5 (Grant et al., 1992), and in two other cases (Chu et al., 1996). Involvement of the sympathetic system is less certain. In the pupil, detection of a sympathetic deficit is problematic if there is coincident parasympathetic damage, because the physiological signs are masked. Furthermore, we consider that topical drug studies may be unreliable in the presence of severe alacrima, because drug penetration into the eye is likely to be greatly enhanced. We found bilateral pupillary redilatation lag without pupillotonia in one patient (proband of family 2), from which we deduce the presence of a sympathetic deficit. Abnormal cardiovascular sympathetic function tests as well as peripheral nerve autonomic abnormalities with cold and dry skin were also seen in our patients (Grant et al., 1992), and postural hypotension in two other cases (Chu et al., 1996). It therefore appears that autonomic neuropathy in this condition is not confined to the lacrymal glands and the upper part of the gastrointestinal tract.
Repeat autonomic function measurements previously had not been made, unlike in two of our patients, who were studied at intervals of 2 and 3 years, respectively. In both patients (5.01 and 3.02), repeat studies indicated a greater degree of orthostatic hypotension, and further blunting of sympathetic vasoconstrictor responses, indicating progression. This would be consistent with the pattern observed in relation to other clinical features, which progress. The case for longitudinal study in these subjects therefore is strong. The lesion site resulting in sympathetic dysfunction is not known, as there are limited neuropathological studies. There may be central or peripheral involvement; if the latter, there may be involvement of autonomic ganglia and the nicotinic acetylcholine receptor, thus affecting both sympathetic and parasympathetic function. It could be said that the abnormal sudomotor tests observed in family 6 indicate a post‐ganglionic sympathetic dysfunction. The lesion may have been to post‐ganglionic pathways at either pre‐ or postsynaptic sites. Further studies, using appropriate physiological and pharmacological tests, should determine the site or sites involved.
Upper and lower motor neurone abnormalities were present to a variable degree of severity in all families. This was also the case for motor neuropathy which was present in the upper and lower limbs on clinical examination and nerve conduction studies. In the upper limbs, there was marked patchiness with selective involvement of the ulnar nerve. In the Portuguese case where the triple A mutation is reported here, there was similar motor neuropathy, most evident on nerve conduction studies (Table 5). In this case, sural nerve biopsy was carried out and nerve pathology on light microscopy was normal. The marked selective involvement of the ulnar nerve is interesting and not seen in hereditary motor and sensory neuropathies. The cause of this patchiness is unknown. As the disease progresses, other nerve involvement becomes apparent, as seen in family 1 where there is both severe ulnar nerve and moderate/severe median nerve motor neuropathy. Nerve conduction studies in our families suggest that the peripheral nerve pathology involves axonal loss. The increased tone, reflexes and abnormal central motor conduction time suggests that there is also involvement of the corticospinal tracts, although MRI of the head and spinal cord was normal. Therefore, sequencing of the triple A gene should be considered in those patients with a complicated axonal neuropathy.
The clinical features of the families with triple A mutations are diverse, involving many systems. This reflects the function of the ALADIN gene which belongs to the WD repeat protein family which exhibits wide functional diversity that includes protein–protein interactions, signal transduction, RNA processing, vesicular trafficking, cytoskeleton assembly and cell division control (Neer et al., 1994; Smith et al., 1999). The families show considerable variability in disease severity that could be due to different mutation types and position. Looking for genotype–phenotype correlations is difficult. Although we have the largest reported series of families with compound heterozygous mutations, no two families have the same two mutations. However, we can look at two broad groups of families. Families 1 and 2 have a later age of onset and less severe clinical phenotype than the other families analysed. They are both compound heterozygotes with a frameshift mutation on one allele and an exon 1 missense mutation on the other. Families 3–5 have a more severe phenotype with a frameshift mutation on one allele and a mutation creating a stop codon change or a mutation in an important functional position on the gene (Ser263→Thr). The mutations in families 3–5 are more likely to lead to a greater loss of function in the triple A gene and hence greater phenotype severity, whereas in families 1 and 2 there is likely to be only partial loss of function. The issue of genotype–phenotype correlations along with the structure and function of the triple A gene need to be analysed further with screening for mutations in families and subsequent protein analysis and disease modelling.
Five families had a total of nine compound heterozygous mutations, and one family (family 6) had two homozygous mutations, although the deletion in the poly(A) tract may not be pathogenic; these changes were spread throughout the triple A gene. Two families had the classical features of triple A syndrome, but no mutations were detected, suggesting genetic heterogeneity. In these two cases, no other family members were available for us to carry out haplotype analysis using markers on chromosome 12q13. Families with mutations had the classical triple A phenotype of achalasia, alacrima, adrenal abnormalities and progressive neurological abnormalities. A number of associated neurological features are described in detail in these families, such as preferential ulnar nerve involvement with distal amyotrophy, widespread autonomic nervous system abnormalities, corticospinal tract damage and cranial nerve palsies. Genotype–phenotype analysis based on these families revealed that frameshift, stop codon and functionally significant mutations are likely to lead to a more severe phenotype, most probably occurring by a loss‐of‐function effect on the protein. Further mutation screening and functional analysis will lead to a greater understanding of this ubiquitous developmental gene.
Acknowledgements
We are grateful to the families for their help. This research was supported by The Wellcome Trust.
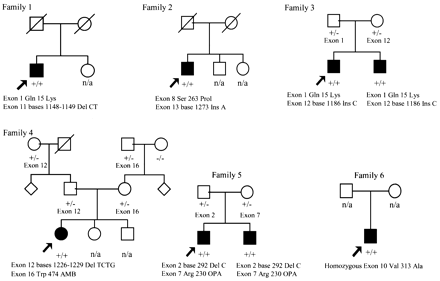
Fig. 1 Family trees of all kindreds with mutations in the triple A gene. Circle = female; square = male; filled = clinically affected. Arrow indicates the proband. +/+ = two mutations; +/– = normal carrier of a mutation; –/– = no mutation; del = deletion; ins = insertion; n/a = not tested genetically. The mutation details are given beneath the affected individual. In the text and subsequent tables when referring to the family number this means the affected individual in the family. In family 3, 3.1 is the proband and 3.2 the other affected sibling, family 5, 5.1 is the proband and 5.2 the other affected sibling. Family members that are heterozygous are indicated with the exonic location of the mutation. All mutations segregate with the disease in an autosomal recessive pattern. AMB = stop codon TAG; OPA = stop codon TGA.
Triple A gene primer sequences
| Exon | Forward (5′–3′) | Reverse (5′–3′) |
| 1 | GGAGTTTGCCGACTGCAGAC | CCTGTCACACTGCCTCCTTTC |
| 2 | GCATTTGAGTTCTATAATAAGGAC | CTCTGGAATCTCTTATACTTAGC |
| 3 | CACTCTGGACACCCACTC | CATAGTTGGCACTCATTAATTG |
| 4 and 5 | AGTAGGAGTCTTTGCCTTCTC | AGAGTGTGGTGTTGAGAGCAC |
| 6 | TCAGGTTCAAGAACTACAGGAC | TTACTGGAAATGAATGTGAGC |
| 7 | CTCCAGATTAGAGTATTCTCAGC | TCCTTAACTGCACTCTGGTC |
| 8 | AGACCTTGCAGATTACCTTC | TCTGGGTAAGTTTAAGACTG |
| 9 | ATTAGAGAGGCCAGCCCACTG | AAGTTGGACCTACCTCCCTTGAC |
| 10 and 11 | AAAGGCACTTAGCTCCTGGAAG | TCTATATTTCCCTTTATCCCTCAGAG |
| 12 and 13 | TTAGGAGATTTCGAGGTGTTGATG | GGCACGGCCTCATTAGATTAAC |
| 14 and 15 | GAGTTCTCCTCTGCCCATGTC | AGAGCCATACAGCAGCCAAG |
| 16 | AGTTGGATGGAGAAGCTGAGG | GCCTTAACCCAAAGTCCATG |
| Exon | Forward (5′–3′) | Reverse (5′–3′) |
| 1 | GGAGTTTGCCGACTGCAGAC | CCTGTCACACTGCCTCCTTTC |
| 2 | GCATTTGAGTTCTATAATAAGGAC | CTCTGGAATCTCTTATACTTAGC |
| 3 | CACTCTGGACACCCACTC | CATAGTTGGCACTCATTAATTG |
| 4 and 5 | AGTAGGAGTCTTTGCCTTCTC | AGAGTGTGGTGTTGAGAGCAC |
| 6 | TCAGGTTCAAGAACTACAGGAC | TTACTGGAAATGAATGTGAGC |
| 7 | CTCCAGATTAGAGTATTCTCAGC | TCCTTAACTGCACTCTGGTC |
| 8 | AGACCTTGCAGATTACCTTC | TCTGGGTAAGTTTAAGACTG |
| 9 | ATTAGAGAGGCCAGCCCACTG | AAGTTGGACCTACCTCCCTTGAC |
| 10 and 11 | AAAGGCACTTAGCTCCTGGAAG | TCTATATTTCCCTTTATCCCTCAGAG |
| 12 and 13 | TTAGGAGATTTCGAGGTGTTGATG | GGCACGGCCTCATTAGATTAAC |
| 14 and 15 | GAGTTCTCCTCTGCCCATGTC | AGAGCCATACAGCAGCCAAG |
| 16 | AGTTGGATGGAGAAGCTGAGG | GCCTTAACCCAAAGTCCATG |
Triple A gene primer sequences
| Exon | Forward (5′–3′) | Reverse (5′–3′) |
| 1 | GGAGTTTGCCGACTGCAGAC | CCTGTCACACTGCCTCCTTTC |
| 2 | GCATTTGAGTTCTATAATAAGGAC | CTCTGGAATCTCTTATACTTAGC |
| 3 | CACTCTGGACACCCACTC | CATAGTTGGCACTCATTAATTG |
| 4 and 5 | AGTAGGAGTCTTTGCCTTCTC | AGAGTGTGGTGTTGAGAGCAC |
| 6 | TCAGGTTCAAGAACTACAGGAC | TTACTGGAAATGAATGTGAGC |
| 7 | CTCCAGATTAGAGTATTCTCAGC | TCCTTAACTGCACTCTGGTC |
| 8 | AGACCTTGCAGATTACCTTC | TCTGGGTAAGTTTAAGACTG |
| 9 | ATTAGAGAGGCCAGCCCACTG | AAGTTGGACCTACCTCCCTTGAC |
| 10 and 11 | AAAGGCACTTAGCTCCTGGAAG | TCTATATTTCCCTTTATCCCTCAGAG |
| 12 and 13 | TTAGGAGATTTCGAGGTGTTGATG | GGCACGGCCTCATTAGATTAAC |
| 14 and 15 | GAGTTCTCCTCTGCCCATGTC | AGAGCCATACAGCAGCCAAG |
| 16 | AGTTGGATGGAGAAGCTGAGG | GCCTTAACCCAAAGTCCATG |
| Exon | Forward (5′–3′) | Reverse (5′–3′) |
| 1 | GGAGTTTGCCGACTGCAGAC | CCTGTCACACTGCCTCCTTTC |
| 2 | GCATTTGAGTTCTATAATAAGGAC | CTCTGGAATCTCTTATACTTAGC |
| 3 | CACTCTGGACACCCACTC | CATAGTTGGCACTCATTAATTG |
| 4 and 5 | AGTAGGAGTCTTTGCCTTCTC | AGAGTGTGGTGTTGAGAGCAC |
| 6 | TCAGGTTCAAGAACTACAGGAC | TTACTGGAAATGAATGTGAGC |
| 7 | CTCCAGATTAGAGTATTCTCAGC | TCCTTAACTGCACTCTGGTC |
| 8 | AGACCTTGCAGATTACCTTC | TCTGGGTAAGTTTAAGACTG |
| 9 | ATTAGAGAGGCCAGCCCACTG | AAGTTGGACCTACCTCCCTTGAC |
| 10 and 11 | AAAGGCACTTAGCTCCTGGAAG | TCTATATTTCCCTTTATCCCTCAGAG |
| 12 and 13 | TTAGGAGATTTCGAGGTGTTGATG | GGCACGGCCTCATTAGATTAAC |
| 14 and 15 | GAGTTCTCCTCTGCCCATGTC | AGAGCCATACAGCAGCCAAG |
| 16 | AGTTGGATGGAGAAGCTGAGG | GCCTTAACCCAAAGTCCATG |
Triple A families mutation details
| Family | Affected | Consanguinity | Ethnic origin | Mutation type | Mutation (allele 1) | Mutation (allele 2) |
| 1 | 1 | No | English | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 11, bases 1148 and 1149 Del CT |
| 2 | 1 | No | English | Compound heterozygote | Exon 8, Ser 263 Pro | Exon 13, base 1273 Ins A |
| 3 | 2 | No | English/German | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 12, base 1186 Ins C |
| 4 | 1 | No | English/Scottish | Compound heterozygote | Exon 12, bases 1226–1229 Del TCTG | Exon 16, Trp 474 AMB |
| 5 | 2 | No | Welsh/English | Compound heterozygote | Exon 2, base 292 Del C | Exon 7, Arg 230 OPA |
| 6 | 1 | No | Portuguese | Homozygote | Exon 10, Val 313 Ala |
| Family | Affected | Consanguinity | Ethnic origin | Mutation type | Mutation (allele 1) | Mutation (allele 2) |
| 1 | 1 | No | English | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 11, bases 1148 and 1149 Del CT |
| 2 | 1 | No | English | Compound heterozygote | Exon 8, Ser 263 Pro | Exon 13, base 1273 Ins A |
| 3 | 2 | No | English/German | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 12, base 1186 Ins C |
| 4 | 1 | No | English/Scottish | Compound heterozygote | Exon 12, bases 1226–1229 Del TCTG | Exon 16, Trp 474 AMB |
| 5 | 2 | No | Welsh/English | Compound heterozygote | Exon 2, base 292 Del C | Exon 7, Arg 230 OPA |
| 6 | 1 | No | Portuguese | Homozygote | Exon 10, Val 313 Ala |
Ins = insertion; Del = deletion; AMB = stop codon (TAG); OPA = stop codon (TGA).
Triple A families mutation details
| Family | Affected | Consanguinity | Ethnic origin | Mutation type | Mutation (allele 1) | Mutation (allele 2) |
| 1 | 1 | No | English | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 11, bases 1148 and 1149 Del CT |
| 2 | 1 | No | English | Compound heterozygote | Exon 8, Ser 263 Pro | Exon 13, base 1273 Ins A |
| 3 | 2 | No | English/German | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 12, base 1186 Ins C |
| 4 | 1 | No | English/Scottish | Compound heterozygote | Exon 12, bases 1226–1229 Del TCTG | Exon 16, Trp 474 AMB |
| 5 | 2 | No | Welsh/English | Compound heterozygote | Exon 2, base 292 Del C | Exon 7, Arg 230 OPA |
| 6 | 1 | No | Portuguese | Homozygote | Exon 10, Val 313 Ala |
| Family | Affected | Consanguinity | Ethnic origin | Mutation type | Mutation (allele 1) | Mutation (allele 2) |
| 1 | 1 | No | English | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 11, bases 1148 and 1149 Del CT |
| 2 | 1 | No | English | Compound heterozygote | Exon 8, Ser 263 Pro | Exon 13, base 1273 Ins A |
| 3 | 2 | No | English/German | Compound heterozygote | Exon 1, Gln 15 Lys | Exon 12, base 1186 Ins C |
| 4 | 1 | No | English/Scottish | Compound heterozygote | Exon 12, bases 1226–1229 Del TCTG | Exon 16, Trp 474 AMB |
| 5 | 2 | No | Welsh/English | Compound heterozygote | Exon 2, base 292 Del C | Exon 7, Arg 230 OPA |
| 6 | 1 | No | Portuguese | Homozygote | Exon 10, Val 313 Ala |
Ins = insertion; Del = deletion; AMB = stop codon (TAG); OPA = stop codon (TGA).
Triple A families non‐neurological clinical details
| Patient | Age of onset (years) | Age at examination (years) | Presenting clinical feature | Achalasia | Adrenal dysfunction | Alacrima |
| 1 | 16 | 48 | Lower limb weakness | Mild | Mild | Mild |
| 2 | 12 | 37 | Lower limb weakness | Mild | Borderline abnormal | Mild |
| 3.1 | 6 | 36 | Alacrima | Mild | Mild | Severe |
| 3.2 | 3 | 39 | Alacrima | Severe | Mild | Moderate |
| 4 | 8 | 18 | Hypoglycaemic seizures | Moderate | Moderate | Moderate |
| 5.1 | 4 | 32 | Hypoglycaemic seizures | Severe | Moderate | Mild |
| 5.2 | 3 | 30 | Alacrima | Moderate | Moderate | Mild |
| 6 | 25 | 35 | Achalasia + lower limb weakness | Mild | Borderline abnormal | Moderate |
| Patient | Age of onset (years) | Age at examination (years) | Presenting clinical feature | Achalasia | Adrenal dysfunction | Alacrima |
| 1 | 16 | 48 | Lower limb weakness | Mild | Mild | Mild |
| 2 | 12 | 37 | Lower limb weakness | Mild | Borderline abnormal | Mild |
| 3.1 | 6 | 36 | Alacrima | Mild | Mild | Severe |
| 3.2 | 3 | 39 | Alacrima | Severe | Mild | Moderate |
| 4 | 8 | 18 | Hypoglycaemic seizures | Moderate | Moderate | Moderate |
| 5.1 | 4 | 32 | Hypoglycaemic seizures | Severe | Moderate | Mild |
| 5.2 | 3 | 30 | Alacrima | Moderate | Moderate | Mild |
| 6 | 25 | 35 | Achalasia + lower limb weakness | Mild | Borderline abnormal | Moderate |
Achalasia criteria: mild = symptoms but no surgery; moderate = symptoms requiring one or two oesphageal dilatation procedures; severe = >3 oesphageal dilatation procedures. Adrenal criteria: borderline abnormal = normal cortisol, raised adrenocorticotrophic hormone, no replacement; mild = required corticosteroid replacement in the past or continued low dose; moderate = continuous need for corticosteroid replacement, required mineralocorticoid in the past. Alacrima criteria: mild = abnormal Schirmer test and symptoms, eye drops as required; moderate = abnormal Schirmer test and symptoms with daily regular treatment; severe = abnormal Schirmer test, severe symptoms, frequent daily treatment, eye/ski goggles.
Triple A families non‐neurological clinical details
| Patient | Age of onset (years) | Age at examination (years) | Presenting clinical feature | Achalasia | Adrenal dysfunction | Alacrima |
| 1 | 16 | 48 | Lower limb weakness | Mild | Mild | Mild |
| 2 | 12 | 37 | Lower limb weakness | Mild | Borderline abnormal | Mild |
| 3.1 | 6 | 36 | Alacrima | Mild | Mild | Severe |
| 3.2 | 3 | 39 | Alacrima | Severe | Mild | Moderate |
| 4 | 8 | 18 | Hypoglycaemic seizures | Moderate | Moderate | Moderate |
| 5.1 | 4 | 32 | Hypoglycaemic seizures | Severe | Moderate | Mild |
| 5.2 | 3 | 30 | Alacrima | Moderate | Moderate | Mild |
| 6 | 25 | 35 | Achalasia + lower limb weakness | Mild | Borderline abnormal | Moderate |
| Patient | Age of onset (years) | Age at examination (years) | Presenting clinical feature | Achalasia | Adrenal dysfunction | Alacrima |
| 1 | 16 | 48 | Lower limb weakness | Mild | Mild | Mild |
| 2 | 12 | 37 | Lower limb weakness | Mild | Borderline abnormal | Mild |
| 3.1 | 6 | 36 | Alacrima | Mild | Mild | Severe |
| 3.2 | 3 | 39 | Alacrima | Severe | Mild | Moderate |
| 4 | 8 | 18 | Hypoglycaemic seizures | Moderate | Moderate | Moderate |
| 5.1 | 4 | 32 | Hypoglycaemic seizures | Severe | Moderate | Mild |
| 5.2 | 3 | 30 | Alacrima | Moderate | Moderate | Mild |
| 6 | 25 | 35 | Achalasia + lower limb weakness | Mild | Borderline abnormal | Moderate |
Achalasia criteria: mild = symptoms but no surgery; moderate = symptoms requiring one or two oesphageal dilatation procedures; severe = >3 oesphageal dilatation procedures. Adrenal criteria: borderline abnormal = normal cortisol, raised adrenocorticotrophic hormone, no replacement; mild = required corticosteroid replacement in the past or continued low dose; moderate = continuous need for corticosteroid replacement, required mineralocorticoid in the past. Alacrima criteria: mild = abnormal Schirmer test and symptoms, eye drops as required; moderate = abnormal Schirmer test and symptoms with daily regular treatment; severe = abnormal Schirmer test, severe symptoms, frequent daily treatment, eye/ski goggles.
Triple A families neurological manifestations
| Patient | Cranial nerve examination | ANS | Distal weakness and wasting | Reflexes | Sensory examination | Pupil diameter | Pupil reactivity |
| 1 | Bilateral OA, bilateral XII, dysarthria | Mild | Severe weakness, mild distal upper and lower limb amyotrophy | Brisk, absent AJ, ExP | Loss of vibration sense | Normal | Pupillotonia, LND |
| 2 | Bilateral OA, R Horner’s, RP | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Loss of vibration sense | Normal | Redilatation lag |
| 3.1 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, EqP | Normal | Normal | Pupillotonia, LND |
| 3.2 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, ExP | Normal | Anisocoria | Pupillotonia, LND |
| 4 | Bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Normal | Normal | Normal |
| 5.1 | Mild bilateral OA | Moderate | Moderate weakness, mild distal upper and lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Miosis | Pupillotonia, no LND |
| 5.2 | Mild bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Normal | Pupillotonia, no LND |
| 6 | Unilateral IX and X, bilateral XI and XII, dysarthria | Moderate | Moderate weakness, symmetrical | Brisk, absent AJ, FP | Normal | Normal | Normal |
| Patient | Cranial nerve examination | ANS | Distal weakness and wasting | Reflexes | Sensory examination | Pupil diameter | Pupil reactivity |
| 1 | Bilateral OA, bilateral XII, dysarthria | Mild | Severe weakness, mild distal upper and lower limb amyotrophy | Brisk, absent AJ, ExP | Loss of vibration sense | Normal | Pupillotonia, LND |
| 2 | Bilateral OA, R Horner’s, RP | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Loss of vibration sense | Normal | Redilatation lag |
| 3.1 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, EqP | Normal | Normal | Pupillotonia, LND |
| 3.2 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, ExP | Normal | Anisocoria | Pupillotonia, LND |
| 4 | Bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Normal | Normal | Normal |
| 5.1 | Mild bilateral OA | Moderate | Moderate weakness, mild distal upper and lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Miosis | Pupillotonia, no LND |
| 5.2 | Mild bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Normal | Pupillotonia, no LND |
| 6 | Unilateral IX and X, bilateral XI and XII, dysarthria | Moderate | Moderate weakness, symmetrical | Brisk, absent AJ, FP | Normal | Normal | Normal |
OA = optic atrophy; RP = retinopathy; ANS = autonomic nervous system abnormalities; AJ = ankle jerks; ExP = extensor plantar reflex; EqP = equivocal plantar reflex; FP = flexor plantar reflex; LND = light‐near dissociation; IX, X, XI and XII = 9th, 10th, 11th and 12th cranial nerves, respectively. Families 1–5 had formal pupillography carried out, family 6 was clinically examined.
Triple A families neurological manifestations
| Patient | Cranial nerve examination | ANS | Distal weakness and wasting | Reflexes | Sensory examination | Pupil diameter | Pupil reactivity |
| 1 | Bilateral OA, bilateral XII, dysarthria | Mild | Severe weakness, mild distal upper and lower limb amyotrophy | Brisk, absent AJ, ExP | Loss of vibration sense | Normal | Pupillotonia, LND |
| 2 | Bilateral OA, R Horner’s, RP | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Loss of vibration sense | Normal | Redilatation lag |
| 3.1 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, EqP | Normal | Normal | Pupillotonia, LND |
| 3.2 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, ExP | Normal | Anisocoria | Pupillotonia, LND |
| 4 | Bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Normal | Normal | Normal |
| 5.1 | Mild bilateral OA | Moderate | Moderate weakness, mild distal upper and lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Miosis | Pupillotonia, no LND |
| 5.2 | Mild bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Normal | Pupillotonia, no LND |
| 6 | Unilateral IX and X, bilateral XI and XII, dysarthria | Moderate | Moderate weakness, symmetrical | Brisk, absent AJ, FP | Normal | Normal | Normal |
| Patient | Cranial nerve examination | ANS | Distal weakness and wasting | Reflexes | Sensory examination | Pupil diameter | Pupil reactivity |
| 1 | Bilateral OA, bilateral XII, dysarthria | Mild | Severe weakness, mild distal upper and lower limb amyotrophy | Brisk, absent AJ, ExP | Loss of vibration sense | Normal | Pupillotonia, LND |
| 2 | Bilateral OA, R Horner’s, RP | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Loss of vibration sense | Normal | Redilatation lag |
| 3.1 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, EqP | Normal | Normal | Pupillotonia, LND |
| 3.2 | Bilateral OA, bilateral XII, dysarthria | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, ExP | Normal | Anisocoria | Pupillotonia, LND |
| 4 | Bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Brisk, absent AJ, FP | Normal | Normal | Normal |
| 5.1 | Mild bilateral OA | Moderate | Moderate weakness, mild distal upper and lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Miosis | Pupillotonia, no LND |
| 5.2 | Mild bilateral OA | Mild | Mild weakness, hypothenar prominent wasting, lower limb amyotrophy | Slightly brisk, absent AJ, FP | Normal | Normal | Pupillotonia, no LND |
| 6 | Unilateral IX and X, bilateral XI and XII, dysarthria | Moderate | Moderate weakness, symmetrical | Brisk, absent AJ, FP | Normal | Normal | Normal |
OA = optic atrophy; RP = retinopathy; ANS = autonomic nervous system abnormalities; AJ = ankle jerks; ExP = extensor plantar reflex; EqP = equivocal plantar reflex; FP = flexor plantar reflex; LND = light‐near dissociation; IX, X, XI and XII = 9th, 10th, 11th and 12th cranial nerves, respectively. Families 1–5 had formal pupillography carried out, family 6 was clinically examined.
Triple A families neurophysiology details
| Patient | Median nerve | Ulnar nerve | |||||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | MNCV (ms) | F wave (m/s) | SAP (mV) | SNCV (ms) | CMAP wrist (mV) | DML (ms) | MNCV (ms) | |
| 1 | Absent | 40 | 2.5 | 5 | 38 | 41.3 | Absent | Absent | Absent | Absent | Absent |
| 2 | 9 | 55.5 | 9.4 | 4 | 54 | 26–37 | 6.5 | 60 | 0.7 | 3.9 | 39 |
| 3.1 | 6 | 50 | 6.5 | 3.4 | 47 | 32.3 | 4 | 47.5 | 0.5 | 3.9 | 36 |
| 3.2 | 22.7 | n/a | 5.3 | 4.5 | 52 | 29.6 | 12.2 | n/a | 4.9 | 5.2 | n/a |
| 4 | 16 | 58.5 | 5.3 | 4.4 | 52 | 29.9 | 7.5 | 61.5 | 1.8 | 4.6 | n/a |
| 6* | 21.0 | 42.5 | 3.7 | 3.6 | 45.8 | 30 | 22.4 | 39.3 | 2.1 | 3.7 | 41.3 |
| Patient | Sural nerve | Common peroneal nerve | EMG | CMCT | |||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | F wave (ms) | Tibialis anterior | ||||||
| 1 | 4 | 47 | 0.013 | 4.5 | n/a | Chronic partial denervation | Prolonged | ||||
| 2 (post tibial) | 7 | 37 | 1.2 | 5.6 | Absent | Chronic partial denervation | Prolonged | ||||
| 3.1 | 2.5 | 37 | 3.5 | 3.9 | Absent | Chronic partial denervation | Normal | ||||
| 3.2 (post tibial) | 6.2 | n/a | 0.4 | 6.5 | 54.8 | n/a | Normal | ||||
| 4 | 17 | 38 | 1.3 | 4.7 | 52.9 | Chronic partial denervation | n/a | ||||
| 5.1 (age 10 years) | 26 | 56 | 4.0 abductor hallucis | ||||||||
| 5.1 (age 17 years) | 15 | 1.3 abductor hallucis | |||||||||
| 5.1 (age 18 years) | 7 | 1.3 abductor hallucis | |||||||||
| 5.2 (age 13 years) | 3 | 0.3 abductor hallucis | |||||||||
| 5.2 (age 16 years) | 3 | 0.1 abductor hallucis | |||||||||
| 6* | 5.0 | 41.3 | 0.3 | 3.9 | Absent | Chronic partial denervation | Prolonged | ||||
| Patient | Median nerve | Ulnar nerve | |||||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | MNCV (ms) | F wave (m/s) | SAP (mV) | SNCV (ms) | CMAP wrist (mV) | DML (ms) | MNCV (ms) | |
| 1 | Absent | 40 | 2.5 | 5 | 38 | 41.3 | Absent | Absent | Absent | Absent | Absent |
| 2 | 9 | 55.5 | 9.4 | 4 | 54 | 26–37 | 6.5 | 60 | 0.7 | 3.9 | 39 |
| 3.1 | 6 | 50 | 6.5 | 3.4 | 47 | 32.3 | 4 | 47.5 | 0.5 | 3.9 | 36 |
| 3.2 | 22.7 | n/a | 5.3 | 4.5 | 52 | 29.6 | 12.2 | n/a | 4.9 | 5.2 | n/a |
| 4 | 16 | 58.5 | 5.3 | 4.4 | 52 | 29.9 | 7.5 | 61.5 | 1.8 | 4.6 | n/a |
| 6* | 21.0 | 42.5 | 3.7 | 3.6 | 45.8 | 30 | 22.4 | 39.3 | 2.1 | 3.7 | 41.3 |
| Patient | Sural nerve | Common peroneal nerve | EMG | CMCT | |||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | F wave (ms) | Tibialis anterior | ||||||
| 1 | 4 | 47 | 0.013 | 4.5 | n/a | Chronic partial denervation | Prolonged | ||||
| 2 (post tibial) | 7 | 37 | 1.2 | 5.6 | Absent | Chronic partial denervation | Prolonged | ||||
| 3.1 | 2.5 | 37 | 3.5 | 3.9 | Absent | Chronic partial denervation | Normal | ||||
| 3.2 (post tibial) | 6.2 | n/a | 0.4 | 6.5 | 54.8 | n/a | Normal | ||||
| 4 | 17 | 38 | 1.3 | 4.7 | 52.9 | Chronic partial denervation | n/a | ||||
| 5.1 (age 10 years) | 26 | 56 | 4.0 abductor hallucis | ||||||||
| 5.1 (age 17 years) | 15 | 1.3 abductor hallucis | |||||||||
| 5.1 (age 18 years) | 7 | 1.3 abductor hallucis | |||||||||
| 5.2 (age 13 years) | 3 | 0.3 abductor hallucis | |||||||||
| 5.2 (age 16 years) | 3 | 0.1 abductor hallucis | |||||||||
| 6* | 5.0 | 41.3 | 0.3 | 3.9 | Absent | Chronic partial denervation | Prolonged | ||||
SAP = sensory nerve action potential; SNCV = sensory nerve conduction velocity; CMAP = compound muscle action potential amplitude; DML = distal motor latency; MNCV = motor nerve conduction velocity; CMCT = central motor conduction time, n/a = not available; *this patient was investigated in another laboratory and sural nerve sensory potential was studied instead of the peroneal nerve sensory potential.
Triple A families neurophysiology details
| Patient | Median nerve | Ulnar nerve | |||||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | MNCV (ms) | F wave (m/s) | SAP (mV) | SNCV (ms) | CMAP wrist (mV) | DML (ms) | MNCV (ms) | |
| 1 | Absent | 40 | 2.5 | 5 | 38 | 41.3 | Absent | Absent | Absent | Absent | Absent |
| 2 | 9 | 55.5 | 9.4 | 4 | 54 | 26–37 | 6.5 | 60 | 0.7 | 3.9 | 39 |
| 3.1 | 6 | 50 | 6.5 | 3.4 | 47 | 32.3 | 4 | 47.5 | 0.5 | 3.9 | 36 |
| 3.2 | 22.7 | n/a | 5.3 | 4.5 | 52 | 29.6 | 12.2 | n/a | 4.9 | 5.2 | n/a |
| 4 | 16 | 58.5 | 5.3 | 4.4 | 52 | 29.9 | 7.5 | 61.5 | 1.8 | 4.6 | n/a |
| 6* | 21.0 | 42.5 | 3.7 | 3.6 | 45.8 | 30 | 22.4 | 39.3 | 2.1 | 3.7 | 41.3 |
| Patient | Sural nerve | Common peroneal nerve | EMG | CMCT | |||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | F wave (ms) | Tibialis anterior | ||||||
| 1 | 4 | 47 | 0.013 | 4.5 | n/a | Chronic partial denervation | Prolonged | ||||
| 2 (post tibial) | 7 | 37 | 1.2 | 5.6 | Absent | Chronic partial denervation | Prolonged | ||||
| 3.1 | 2.5 | 37 | 3.5 | 3.9 | Absent | Chronic partial denervation | Normal | ||||
| 3.2 (post tibial) | 6.2 | n/a | 0.4 | 6.5 | 54.8 | n/a | Normal | ||||
| 4 | 17 | 38 | 1.3 | 4.7 | 52.9 | Chronic partial denervation | n/a | ||||
| 5.1 (age 10 years) | 26 | 56 | 4.0 abductor hallucis | ||||||||
| 5.1 (age 17 years) | 15 | 1.3 abductor hallucis | |||||||||
| 5.1 (age 18 years) | 7 | 1.3 abductor hallucis | |||||||||
| 5.2 (age 13 years) | 3 | 0.3 abductor hallucis | |||||||||
| 5.2 (age 16 years) | 3 | 0.1 abductor hallucis | |||||||||
| 6* | 5.0 | 41.3 | 0.3 | 3.9 | Absent | Chronic partial denervation | Prolonged | ||||
| Patient | Median nerve | Ulnar nerve | |||||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | MNCV (ms) | F wave (m/s) | SAP (mV) | SNCV (ms) | CMAP wrist (mV) | DML (ms) | MNCV (ms) | |
| 1 | Absent | 40 | 2.5 | 5 | 38 | 41.3 | Absent | Absent | Absent | Absent | Absent |
| 2 | 9 | 55.5 | 9.4 | 4 | 54 | 26–37 | 6.5 | 60 | 0.7 | 3.9 | 39 |
| 3.1 | 6 | 50 | 6.5 | 3.4 | 47 | 32.3 | 4 | 47.5 | 0.5 | 3.9 | 36 |
| 3.2 | 22.7 | n/a | 5.3 | 4.5 | 52 | 29.6 | 12.2 | n/a | 4.9 | 5.2 | n/a |
| 4 | 16 | 58.5 | 5.3 | 4.4 | 52 | 29.9 | 7.5 | 61.5 | 1.8 | 4.6 | n/a |
| 6* | 21.0 | 42.5 | 3.7 | 3.6 | 45.8 | 30 | 22.4 | 39.3 | 2.1 | 3.7 | 41.3 |
| Patient | Sural nerve | Common peroneal nerve | EMG | CMCT | |||||||
| SAP (mV) | SNCV (ms) | CMAP (mV) | DML (ms) | F wave (ms) | Tibialis anterior | ||||||
| 1 | 4 | 47 | 0.013 | 4.5 | n/a | Chronic partial denervation | Prolonged | ||||
| 2 (post tibial) | 7 | 37 | 1.2 | 5.6 | Absent | Chronic partial denervation | Prolonged | ||||
| 3.1 | 2.5 | 37 | 3.5 | 3.9 | Absent | Chronic partial denervation | Normal | ||||
| 3.2 (post tibial) | 6.2 | n/a | 0.4 | 6.5 | 54.8 | n/a | Normal | ||||
| 4 | 17 | 38 | 1.3 | 4.7 | 52.9 | Chronic partial denervation | n/a | ||||
| 5.1 (age 10 years) | 26 | 56 | 4.0 abductor hallucis | ||||||||
| 5.1 (age 17 years) | 15 | 1.3 abductor hallucis | |||||||||
| 5.1 (age 18 years) | 7 | 1.3 abductor hallucis | |||||||||
| 5.2 (age 13 years) | 3 | 0.3 abductor hallucis | |||||||||
| 5.2 (age 16 years) | 3 | 0.1 abductor hallucis | |||||||||
| 6* | 5.0 | 41.3 | 0.3 | 3.9 | Absent | Chronic partial denervation | Prolonged | ||||
SAP = sensory nerve action potential; SNCV = sensory nerve conduction velocity; CMAP = compound muscle action potential amplitude; DML = distal motor latency; MNCV = motor nerve conduction velocity; CMCT = central motor conduction time, n/a = not available; *this patient was investigated in another laboratory and sural nerve sensory potential was studied instead of the peroneal nerve sensory potential.
References
Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production.
Beaudoing E, Freier S, Wyatt JR, Claverie J‐M, Gautheret D. Patterns of variant polyadenylation signal usage in human genes.
Bentes C, Santos‐Bento M, de Sa J, de Lurdes Sales Luis M, de Carvalho M. Allgrove syndrome in adulthood.
Chu ML, Berlin D, Axelrod FB. Allgrove syndrome: documenting cholinergic dysfunction by autonomic tests.
Clark AJ, Weber A. Adrenocorticotropin insensitivity syndromes. [Review].
De Lisa JA, Lee HJ, Baran EM, Lai K‐A, Spielholtz N, MacKenzie K. Manual of nerve conduction velocity and clinical neurophysiology, 3rd edn. New York: Raven Press;
Dumic M, Radica A, Sabol Z, Plavsic V, Brkljacic L, Sarnavka V, et al. Adrenocorticotropic hormone insensitivity associated with autonomic nervous system disorders.
Ehrich E, Aranoff G, Johnson WG. Familial achalasia associated with adrenocortical insufficiency, alacrima, and neurological abnormalities.
Gazarian M, Cowell CT, Bonney M, Grigor WG. The ‘4A’ syndrome: adrenocortical insufficiency associated with achalasia, alacrima, autonomic and other neurological abnormalities.
Geffner ME, Lippe BM, Kaplan SA, Berquist WE, Bateman JB, Paterno VI, et al. Selective ACTH insensitivity, achalasia, and alacrima: a multisystem disorder presenting in childhood.
Grant DB, Dunger DB, Smith I, Hyland K. Familial glucocorticoid deficiency with achalasia of the cardia associated with mixed neuropathy, long‐tract degeneration and mild dementia.
Grant DB, Barnes ND, Dumic M, Ginalska‐Malinowska M, Milla PJ, von Petrykowski W, et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome.
Hadj‐Rabia S, Salomon R, Pelet A, Penet C, Rotschild A, de Laet MH, et al. Linkage disequilibrium in inbred North African families allows fine genetic and physical mapping of triple A syndrome.
Hammami A, Trabelsi M, Bennaceur B, Boukhris R. Association d’une maladie d’Addison, d’une achalasie du cardia et d’une alacrymation. A propos de deux observations. [Review].
Handschug K, Sperling S, Yoon SJ, Hennig S, Clark AJ, Huebner A. Triple A syndrome is caused by mutations in AAAS, a new WD‐repeat protein gene.
Heinrichs C, Tsigos C, Deschepper J, Drews R, Collu R, Dugardeyn C, et al. Familial adrenocorticotropin unresponsiveness associated with alacrima and achalasia: biochemical and molecular studies in two siblings with clinical heterogeneity.
Hübschmann K. Achalasie, Alakrimie und Cortisolmangel—das Allgrove Syndrom. [Review].
Huebner A, Elias LL, Clark AJ. ACTH resistance syndromes. [Review].
Huebner A, Yoon SJ, Özkinay F, Hilscher C, Lee H, Clark AJ, et al. Triple A syndrome—clinical aspects and molecular genetics.
Lee H, Choi E, Seomun Y, Montgomery K, Huebner A, Lee E, et al. High‐resolution transcript map of the region spanning D12S1629 and D12S312 at chromosome 12q13: triple A syndrome‐linked region.
Mathias CJ, Bannister R. Investigation of autonomic disorders. In: Mathias CJ, Bannister R, editors. Autonomic failure: a textbook of clinical disorders of the autonomic nervous system. 4th edn. Oxford: Oxford University Press;
Moore PS, Couch RM, Perry YS, Shuckett EP, Winter JS. Allgrove syndrome: an autosomal recessive syndrome of ACTH insensitivity, achalasia and alacrima.
Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory‐protein family of WD‐repeat proteins. [Review].
Sandrini F, Farmakidis C, Kirschner LS, Wu SM, Tullio‐Pelet A, Lyonnet S, et al. Spectrum of mutations of the AAAS gene in Allgrove syndrome: lack of mutations in six kindreds with isolated resistance to corticotropin.
Smith SA, Smith SE. Bilateral Horner’s syndrome: detection and occurrence.
Smith SE, Smith SA, Brown PM, Fox C, Sonksen PH. Pupillary signs in diabetic autonomic neuropathy.
Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. [Review].
Stratakis CA, Lin JP, Pras E, Rennert OM, Bourdony CJ, Chan WY. Segregation of Allgrove (triple‐A) syndrome in Puerto Rican kindreds with chromosome 12 (12q13) polymorphic markers.
Stuckey BG, Mastaglia FL, Reed WD, Pullan PT. Glucocorticoid insufficiency, achalasia, alacrima with autonomic motor neuropathy.
Tullio‐Pelet A, Salomon R, Hadj‐Rabia S, Mugnier C, de Laet MH, Chaouachi B, et al. Mutant WD‐repeat protein in triple‐A syndrome.
Weber A, Wienker TF, Jung M, Easton D, Dean HJ, Heinrichs C, et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster.
Yuregir GT, Aksoy K, Curuk MA, Dikmen N, Fei YJ, Baysal E, et al. Hb H disease in a Turkish family resulting from the interaction of a deletional alpha‐thalassaemia‐1 and a newly discovered poly A mutation.


{kind=link}