




Abstract
A neurological syndrome involving progressive action tremor with ataxia, cognitive decline and generalized brain atrophy has been described recently in some adult males with pre‐mutation alleles of the fragile X syndrome (FXS) fragile X mental retardation gene (FMR1). Neurohistological studies have now been performed on the brains of four elderly premutation carriers, not reported previously, who displayed the neurological phenotype. Eosinophilic, intranuclear inclusions were present in both neuronal and astrocytic nuclei of the cortex in all four individuals. Systematic analysis of the brains of two of these carriers demonstrated the presence of the intranuclear inclusions throughout the cerebrum and brainstem, being most numerous in the hippocampal formation. The cerebellum displayed marked dropout of Purkinje cells, Purkinje axonal torpedoes and Bergmann gliosis. Intranuclear inclusions were absent from Purkinje cells, although they were present in a small number of neurones in the dentate nucleus and diffusely in cerebellar astrocytes. The presence of inclusions in the brains of all four FXS carriers with the neurological findings provides further support for a unique clinical entity associated with pre‐mutation FMR1 alleles. The origin of the inclusions is unknown, although elevated FMR1 mRNA levels in these pre‐mutation carriers may lead to the neuropathological changes.
Introduction
Fragile X syndrome (FXS) is the most common inherited form of mental retardation, with carrier frequencies estimated to be one in 260 females and one in 760 males (Rousseau et al., 1995, 1996). FXS is caused by a trinucleotide repeat (CGG) expansion in the fragile X mental retardation 1 gene (FMR1) (Verkerk et al., 1991; Yu et al., 1991). For alleles exceeding 200 repeats (full mutation range), the promoter region of the FMR1 gene generally becomes hypermethylated, with consequent transcriptional silencing (Fu et al., 1991; Oberlé et al., 1991; Pieretti et al., 1991; Snow et al., 1993). Individuals with 55–200 CGG repeats (pre‐mutation carriers) typically do not show the full FXS phenotype, although a subgroup does display some physical features of FXS (Dorn et al., 1994; Loesch et al., 1994; Smits et al., 1994; Hagerman et al., 1996; Riddle et al., 1998; Tassone et al., 2000c) and/or mild cognitive and emotional problems (Hagerman et al., 1996; Franke et al., 1998; Tassone et al., 2000c).
There do appear to be some distinguishing clinical features of pre‐mutation carriers. In particular, premature menopause, present in ∼16–24% of females with pre‐mutation alleles, is considered a phenotype unique to the pre‐mutation (Allingham‐Hawkins et al., 1999). Men with alleles in the low pre‐mutation range (55–100 repeats) have FMR1 mRNA levels that are 2–4 times higher than normal, despite near‐normal levels of fragile X mental retardation 1 gene protein (FMRP); whereas men with 100–200 CGG repeats have mRNA levels that are 4–10 times normal, despite mildly reduced FMRP levels (Tassone et al., 2000a, b). Elevated mRNA levels are also observed for females with pre‐mutation alleles (Tassone et al., 2000b). Thus, elevated FMR1 mRNA represents a molecular phenotype for individuals with pre‐mutation alleles. The elevated mRNA levels are thought to be secondary to a reduced translational efficiency of the FMR1 message, which begins in the premutation range.
We have recently reported another collection of clinical features, which includes progressive intention tremor, ataxia and cognitive decline, that appears to be associated with pre‐mutation alleles in some males older than 50 years (Hagerman et al., 2001). Associated findings include impotence, peripheral neuropathy, mood lability and mild Parkinsonian features. These findings are associated consistently with global reductions of brain volume. All of the adults studied thus far have CGG expansions in the 80–135 CGG repeat range and have elevated (2–4‐fold) FMR1 mRNA levels.
To better understand the neuropathology of this tremor/ataxia syndrome, we have examined the brains of two pre‐mutation males who died with symptoms of this syndrome. Both brains revealed the presence of intranuclear inclusions in both neurones and astroglial cells, with widespread distribution throughout the cerebrum and subcortical regions. Evidence of cerebellar neurodegeneration was present in both brains, as evidenced by Purkinje cell loss and torpedo formation (proximal Purkinje cell axonal swelling indicative of degeneration), and, in one case, spongiosis of cerebellar white matter. More limited analysis of samples from two additional brains of older males with pre‐mutation alleles and neurological symptoms consistent with the tremor/ataxia syndrome (Hagerman et al., 2001) also revealed ubiquitin‐positive intranuclear inclusions.
Case reports
Case 1
The patient in Case 1 died on his 70th birthday in a nursing home. He was a carrier of the FMR1 premutation with 135 CGG repeats both in the blood and throughout the brain. The FMR1 mRNA level was 3.77 (±0.13) times normal in blood. The percentage of FMRP‐positive lymphocytes was 62, which is slightly low. Both of his daughters are carriers of the pre‐mutation, and he has grandchildren with FXS (Fig. 1A). He did well in school and attended college for 4 years. He was a teacher for 5 years in his early career. He had a history of shyness and was a solitary individual throughout his life. He was an insurance adjuster and retired at age 51 years, following a myocardial infarction. An angiogram demonstrated blockage in two coronary arteries, which was treated with nitroglycerin.
His family noticed forgetfulness for current activities, in his early sixties. At age 60, he began falling every 4–6 months due to ataxia and, by age 62, he fell on a weekly basis. His handwriting was tremulous and difficult by age 62. By age 63, he began using a cane for walking and had diminished sensation in the lower extremities. He was diagnosed with diabetes at 63 and began using insulin by 64. At that time, he could not recover from a fall without assistance. By age 65, he was using a walker and his long‐term memory was poor. At 65.5 years, he was admitted to a nursing home, where he became wheelchair‐bound. At age 67, he confused the identity of family members. He subsequently had difficulty in swallowing his food, and choked frequently. He died of pneumonia. Spinocerebellar ataxia types 1, 2, 3, 6 and 7 and Huntington’s disease were ruled out by genetic analysis.
Case 2
This patient died at the age of 78 years. He was a pre‐mutation carrier with 80 CGG repeats. He and his family have been reported previously (Fig. 1B) (Kirkilionis et al., 1992; Mogk et al., 1998). He worked as a grain farmer and was a part‐time minister in his church. At 65 years of age, he began having gait problems with frequent falling, which progressed in severity. He also had poor balance, micrographia and an action tremor in his hands, with some tremor at rest. His memory and other cognitive functions showed a progressive decline.
Neurological examination 5 years before death revealed mild memory dysfunction; slow, clear speech; normal facial expression; bilateral resting hand tremor; mild cogwheeling at both wrists; impaired rapid alternating movements in both upper extremities; wide‐based, slow gait, without retropulsion or propulsion; and impaired tandem gait. CT scans showed mild cortical and cerebellar atrophy. His neurologist felt that he might have had an atypical form of Parkinson’s disease, with both extrapyramidal and cerebellar ataxia components. He eventually lost the ability to walk, talk and swallow. He died in a nursing home. Both his half‐brother (Fig. 1B, III‐3) and mother (II‐2) had similar progressive cognitive declines with ataxia, but autopsies were not performed.
Case 3
In Case 3, the patient died of accidental drowning at 69 years. He was a premutation carrier with 80 CGG repeats. His daughter is also a carrier of the premutation and his grandchildren are affected with FXS. His intelligence was normal and his education included a 4‐year degree in engineering. He was prone to anger outbursts throughout life, though they seemed to improve with age. At age 64, he began suffering from gait instability, with increasing ambulation difficulties and frequent falls. At age 68, a routine colonoscopy discovered a transmural in situ colon cancer, which was surgically resected; this was followed by adjuvant chemotherapy. During the following year, a brain MRI was ordered to evaluate progressive gait ataxia and upper and lower limb dysmetria. The MRI ruled out cerebellar metastatic disease, but revealed global brain atrophy. Cognition and short‐term memory declined in his last years.
Case 4
This patient died at 84 years of chronic congestive heart failure. He was a premutation carrier with 71 CGG repeats. His daughter is also a carrier of the premutation, and his grandchildren are affected with FXS. He developed intention tremor and gait instability in his early 70s and also experienced resting tremor of the jaw. His ambulation declined progressively. At 82 years, he needed a cane, followed by a walker and then a wheelchair at 83 years.
A CT scan was performed at the age of 78, and demonstrated moderate atrophy with prominence of sulci and dilatation of the lateral ventricles. At 80 years old, a neurological exam was performed. It revealed mild hypomimia and bradykinesia; cerebellar testing showed mild dysmetria and slowed alternating movements. Sensory testing was normal. He had long‐standing mitral valve disease and underwent two heart valve replacements at the age of 79 (pig) and 82 years (mechanical). A pacemaker was implanted during the same period. He was diagnosed with Parkinson’s disease, and carbidopa/levodopa (25/100 mg three times daily) improved the condition for the subsequent 4 years. He did not show resting tremor while on carbidopa/levodopa, but jaw tremor was apparent when the medication was discontinued; short‐term memory loss was noted in the last 2 years of life. His writing became progressively smaller to a point where it was difficult to read. Dysphagia developed in his last year of life.
Methods
Autopsy
Autopsy on Case 1 was limited to examination of the brain, which was removed within 10 h of death. The fresh weight of the brain was not obtained. Portions of the right cerebral hemisphere and cerebellum were snap‐frozen in liquid nitrogen and subsequently stored at –70°C. The left hemisphere, including the midbrain and rostral pons, was fixed in 4% paraformaldehyde, and weighed 540 g.
The autopsy on Case 2 was limited to the brain and spinal cord, which were removed within 18 h after death. Fresh brain weight was 1340 g. The right hemisphere, brainstem and cerebellum were formalin‐fixed, and the left hemisphere was frozen.
In both cases, fixed tissue was processed in standard fashion for paraffin embedding. In Case 1, cerebellar samples were limited to small fragments of previously frozen tissue.
Immunochemistry
Histological sections were stained by standard immunochemical techniques. After mounting 4 µm thick sections on poly‐l‐lysine‐coated slides, sections were processed on a Dako Autostainer. The sections were de‐paraffinized, treated with methanol/H2O2 (40%/1% in phosphate‐buffered saline) for 5 min, blocked with Dako peroxidase blocking reagent for 5 min, then incubated with antiserum for 30 min at room temperature. Antibodies and dilutions were as follows: anti‐ubiquitin antibody (Dako, Carpenteria, CA, USA), 1 : 800; anti‐GFAP (glial fibrillary acidic protein) antibody (Dako, Carpenteria, Ca, USA), 1 : 16 000; anti‐polyglutamine antibody (mAB1C2: MAB1574, Chemicon International, Temecula, CA, USA), 1 : 1000. Antibody labelling was visualized using the Envision system (Dako) and horseradish peroxidase for 30 min, and treated with Dako DAB substrate–chromagen solution for 10 min. Sections were dehydrated through graded ethanolic solutions and xylene, and were viewed using a conventional light microscope. Antibodies were diluted by 50% for previously frozen cerebellum to achieve staining results comparable with paraformaldehyde‐fixed tissues.
Electron microscopy
Samples of fixed tissue (1 mm3) were processed in standard fashion for ultrastructural analysis. The hippocampus was examined in Case 1. The frontal lobe and cerebellar white matter were analysed in Case 2. Thin sections of previously selected cells containing inclusions were viewed with either a Phillips CM120 or a JEOL 1010 transmission electron microscope.
Intranuclear inclusion and cell counts
Neurones, astrocytic nuclei and intranuclear inclusions were counted manually in multiple fields, on anti‐ubiquitin‐stained slides at 400×, by a trained technician using an optical analyser system. Counts were made in multiple areas of the brain (Table 1). Overall assessments of cortical sections by two neuropathologists did not reveal evidence of neuronal cell loss other than Purkinje cells, nor was gliosis such as could be expected with neuronal loss noted in grey matter. Rigorous morphometric analysis to quantify neuronal cell loss was considered beyond the scope of this work and was not performed.
DNA analysis
Genomic DNA was isolated from 5 ml of peripheral blood leukocytes using standard methods (Puregene kit; Gentra Inc., Minneapolis, MN, USA). For Southern blot analysis, 5 ml of DNA was digested with EcoRI and NruI; the blots were probed with digoxigenin‐labelled probe (STB12.3). For PCR (polymerase chain reaction) analysis, primers 1 and 3 were used (Brown et al., 1993).
Quantification of FMR1 mRNA levels
Total RNA was prepared from 3 ml of whole blood using the Purescript kit (Gentra Inc.). Reverse transcriptase reactions were performed as described (Tassone et al., 2000b). mRNA levels were determined using the method of quantitative fluorescence reverse transcription–PCR (Tassone et al., 2000b). All quantifications of FMR1 mRNA were performed using a 7700 sequence detector (PE Biosystems) (Livak et al., 1995; Heid et al., 1996), which determines the relative abundances of mRNA using real‐time fluorescence detection of a dual‐labelled (TaqMan) probe (5′ FAM‐oligodeoxynucleotide‐TAMRA 3′) that is complementary to the region being amplified (amplicon). In the current work, the FMR1 amplicon is 122 bp, and spans the junction between exons 3 and 4 of the FMR1 gene (GenBank accession no. L29704). The reference (β‐glucoronidase; GUS) amplicon is 81 bp (GenBank accession no. NM000181), and spans exons 11 and 12 of the GUS gene.
FMRP analysis
The percentage of lymphocytes expressing FMRP was determined by immunocytochemistry of blood smears (Livak et al., 1995; Willemsen et al., 1995, 1997; Heid et al., 1996; Tassone et al., 1999), using the mouse anti‐FMRP monoclonal antibody from hybridoma clone 1C3‐a (Devys et al., 1993).
Neuropathology
Case 1
Gross examination. External examination of the fixed left hemisphere revealed moderate diffuse cortical atrophy, most prominent in the frontal lobes. Coronal sections of the brain showed moderate ventricular dilatation and no abnormalities of deep grey matter structures. The substantia nigra was well pigmented. Sections were taken from the superior and middle frontal gyri, cingulate gyrus, insular cortex, basal ganglia, thalamus, mammillary bodies, superior and middle temporal gyri, amygdaloid nucleus, hippocampus, pre‐ and postcentral gyri, inferior parietal lobule, calcarine cortex, midbrain and rostral pons.
Microscopic examination. Haematoxylin and eosin (H&E)‐stained sections of cerebrum and brainstem revealed the presence of single (rarely double) intranuclear inclusions in neurones and astrocytes (Fig. 2A and B). The inclusions ranged in size from 2 to 5 µm, and were homogeneous, eosinophilic and refractile in appearance. Counts for various regions of the brain are presented in Table 1. The inclusions did not distort the nuclear shape; although, especially in astrocytic nuclei, the nuclear chromatin was clear around the inclusions, giving them a haloed appearance. While neuronal nuclei were not enlarged, astrocytic nuclei were up to twice normal in diameter and were often convoluted in white matter. Intranuclear inclusions were present diffusely and were easily identified in astrocytic nuclei. No inclusions were observed in other glial cell populations. There was neither evident neuronal cell loss nor reactive astrocytosis in cortical grey matter. Neither Lewy bodies nor loss of pigmented cells was observed in the substantia nigra. Examination also revealed Purkinje cell dropout in excess of that seen with ageing alone, Bergmann gliosis and scattered axonal torpedoes (Fig. 2C). Intranuclear inclusions were present infrequently in dentate nucleus neurones, diffusely observed in astroglial nuclei, but not found in Purkinje cells. Bielschowsky staining of the cerebral cortical sections showed age‐appropriate changes, including sparse plaques, rare neurofibrillary tangles without plaque formation in the hippocampus, and lack of stained inclusions (not shown). LFB‐PAS (Luxol fast blue–periodic acid–Schiff) showed negative staining of intranuclear inclusions and no loss of myelin.
Immunochemical staining. Staining with anti‐GFAP antibody revealed scattered reactive astrocytes in the cerebral cortical molecular layer and mild subpial gliosis. Reactive astrocytes were otherwise rare in grey matter and non‐existent in white matter of the cerebrum. In the cerebellum, anti‐GFAP antibody demonstrated Bergmann gliosis, scant reactive astrocytes in white matter and prominent astrogliosis of the dentate nucleus. Anti‐ubiquitin antibody clearly marked intranuclear inclusions in both neurones and astrocytes of the cerebrum and brainstem (Fig. 2E), in occasional neurones of the dentate nucleus and, diffusely, in cerebellar astroglia. Anti‐neurofilament antibody confirmed the presence of dystrophic axons (Fig. 2C). Anti‐polyglutamine antibody showed no staining of intranuclear inclusions (not shown).
Electron microscopy. Intranuclear inclusions in neurones and astrocytes had a similar ultrastructural appearance as compact, non‐membrane bound collections of granulofilamentous material (Fig. 3).
Case 2
Gross examination. On external examinations, the formalin‐fixed right hemisphere revealed diffuse, mild cortical atrophy. Coronal sections displayed an equivocal decrease in the size of the basal ganglia and thalamus. Moderate ventriculomegaly was present. Transverse sections of the brainstem and cerebellum were normal in appearance. The spinal cord was mildly atrophied in the cervical and lumbar enlargements, but showed no abnormalities on transverse sectioning. Brain sections sampled included frontal and occipital cortices, basal ganglia, thalamus, hippocampus, midbrain, pons, medulla, cerebellum and spinal cord.
Microscopic examination. H&E‐stained sections of cortex, basal ganglia and thalamus exhibited discrete eosinophilic intranuclear inclusions in neurones. Inclusions were most abundant in the hippocampus. Cortical counts were made in medium to large pyramidal cells of layers 3–5 that could be identified unambiguously as neurones. Mild vascular hyalinosis was seen in the lenticular nuclei. The substantia nigra showed no loss of pigmented neurones, nor were Lewy bodies present; although intranuclear inclusions were present within pigmented neurones (Table 1). There were rare degenerating neurones as well as a mild, patchy dropout of neurones in the inferior olivary nucleus. The section of cerebellum displayed mild, patchy Purkinje cell dropout and minimal Bergmann gliosis; rare swollen axons were seen in the internal layer on H&E staining and by immunochemical staining (described below). Subfoliar white matter of the cerebellum showed striking spongiosis with rare axonal spheroids (Fig. 4A), which spared perivascular areas and was without inflammatory infiltrates. There was no loss of oligodendroglia and no reactive astrocytosis. The dentate nucleus exhibited no apparent loss of neurones, and only rare neuronal intranuclear inclusions. No intranuclear inclusions were seen in neurones of the spinal cord, though they were present in astroglial nuclei. Bielschowsky stains showed mild, age‐appropriate changes in the cortex, and mild loss of axons in spongiotic areas of cerebellar white matter (Fig. 4B). Solochrome cyanine/H&E and LFB‐PAS staining of cerebellar white matter displayed myelin pallor and vacuolation (Fig. 4C). Intranuclear inclusions did not stain with modified Bielschowsky silver stain or PAS, and did not polarize with Congo red staining.
Immunohistochemical staining. Staining with anti‐ubiquitin antibody demonstrated positive staining in intranuclear inclusions (Fig. 2D). Inclusions were negative for staining with anti‐tau, anti‐cytokeratin, anti‐desmin, anti‐αB crystallin, anti‐vimentin, anti‐GFAP and anti‐neurofilament antibodies; the latter antibody identified swollen axons in the granular cell layer of the cerebellum.
Electron microscopy. Intranuclear inclusions appeared as straight (non‐helical) filaments in haphazard arrangement, identical in appearance to those observed for Case 1 in Fig. 3. The filaments measured 12–15 nm in width. There were no abnormalities of myelin that could not be attributed to post‐mortem artefact.
Case 3
Neuropathological examination was limited to a single small sample of previously frozen cortex. Refractile, eosinophilic, ubiquitin‐positive inclusions similar in appearance to those described above were identifiable. However, due to freezing artefact, more precise analysis was not possible.
Case 4
The entire brain was received fixed in formalin (weight = 1400 g) after prior freezing and thawing. On gross examination, the brain tissue was friable in many places, but histological preparations were suitable for cell identification with histological and immunochemical stains. Preliminary studies showed neuronal and astroglial intranuclear inclusions similar in appearance to those described in Cases 1 and 2. These inclusions were also ubiquitin‐positive in sections of hippocampus and midbrain. The substantia nigra possessed numerous intracytoplasmic Lewy bodies, which were also ubiquitin‐positive, but morphologically distinct from the intranuclear inclusions (Fig. 4D). A section of cerebellum demonstrated astroglia with ubiquitin‐positive intranuclear inclusions, and rare torpedoes with anti‐neurofilament antibody in the granular cell layer. Further analysis of this brain is in progress.
Discussion
Association of intranuclear inclusions with CGG repeat expansions
Neuropathological analysis was performed on brain tissue from four male carriers of premutation alleles of the FMR1 gene. These individuals had all experienced a progressive neurological condition involving action tremor and/or gait ataxia. All four cases had cognitive changes, three with significant cognitive decline and a fourth with memory deficits. For the three cases that were examined by MRI, all showed significant brain atrophy. All four brains possessed significant numbers of intranuclear inclusions in both neuronal and astrocytic cell types, suggesting that such inclusions are a common neuropathological correlate of the tremor/ataxia phenotype among carriers of the FMR1 pre‐mutation.
This is the first description of intranuclear inclusions associated with CGG repeat expansions. It is noteworthy that previous neuropathological investigations of individuals with full mutation alleles have not seen such inclusions (Rudelli et al., 1985; Hinton et al., 1991; Reyniers et al., 1999; Irwin et al., 2000, 2001; Sabaratnam, 2000). Two of these studies included males over 60 years of age (Rudelli et al., 1985; Sabaratnam, 2000). There are some parallels between the neuroanatomical changes in the premutation patients with the tremor/ataxia syndrome and the full mutation patients. Sabaratnam (2000) observed Purkinje cell loss and Bergmann gliosis in an older male with the full mutation. Hypoplasia of the posterior cerebellar vermis in those with the full mutation has also been seen in both males and females (Reiss et al., 1991a, b; Mostofsky et al., 1998). There is also evidence of progressive ventricular dilation (Reiss et al., 1995) and more rapid temporal lobe shrinkage, with age, in those with the full mutation compared with controls (Reiss et al., 1994). The main neuroanatomical differences between those with the full mutation and the premutation patients described in the current work is that overall brain volume tends to be increased in the full mutation (Reiss et al., 1994; Schapiro et al., 1995), and intranuclear inclusions have not been observed. This newly identified syndrome, and its associated neuropathology, is thus distinct from FXS, from both the clinical and neuropathological standpoint. It is not known at present whether female carriers of premutation alleles are also at risk for developing the tremor/ataxia syndrome with inclusions, although epidemiological studies are currently underway to address this issue.
The origin of the intranuclear inclusions is unknown at present. Unlike the CAG repeat disorders, the FMRP sequence is normal, both in premutation alleles and in full mutation patients when detectable, since the CGG expansion resides in the 5′‐untranslated region of FMR1 mRNA. The reactivity of the inclusions to anti‐ubiquitin antibodies suggests that they are somehow related to protein degradation via the proteasomal pathway, although it is not clear which proteins are being targeted. The ubiquitin‐positive feature of the inclusions is shared with the CAG repeat expansion (polyglutamine) disorders, including Huntington’s disease, spinobulbar muscular atrophy, dentatorubral pallidoluysian atrophy and a number of the spinocerebellar ataxias (Robitaille et al., 1997; Evert et al., 2000; Yamada et al., 2000). Moreover, the inclusions seen in the current cases appear morphologically identical to those described in the CAG repeat disorders, and show some similarities in staining characteristics: they are negative for PAS and silver staining and are ubiquitin positive. However, unlike the CAG repeat disorders, the inclusions reported in the current work do not stain for polyglutamine‐containing proteins or peptides. Intranuclear inclusions, primarily in neurones, are also present in neuronal intranuclear inclusion disease, a rare multisystem neurodegenerative disorder seen in young adults. Inclusions in neuronal intranuclear inclusion disease seem to vary in appearance ultrastructurally from that of non‐membrane bound, randomly oriented 8–10 nm filaments (Takahashi et al., 2000) to non‐membrane bound parallel arrays of 8–16 nm filaments that more closely resemble rodlets of Roncoroni (Malandrini et al., 1998) (see below). Kakita and co‐workers (Kakita et al., 1997; Kakita and Takahashi, 1999) reported the presence of ubiquitin‐positive intranuclear inclusions in one patient with sporadic amyotrophic lateral sclerosis without mental abnormalities; those inclusions were limited to the hippocampal pyramidal neurones. While those inclusions were negative for multiple cytoskeletal proteins, staining with anti‐polyglutamine antibody was not reported.
The association of inclusions with the FMR1 gene, which does not produce an abnormal protein product (unlike the polyglutamine products of the CAG disorders), suggests that inclusion formation may be a more general response to cytopathic processes. Although FMRP levels are near normal in the blood of individuals with the tremor/ataxia syndrome, FMR1 mRNA levels typically are elevated by 2–4‐fold. Thus, it is possible that the elevated mRNA levels per se may have a cumulative cytotoxic effect that may lead to inclusion formation. In this regard, Liquori et al. (2001) recently reported that small intranuclear foci are present in myotonic dystrophy type I (DMPK gene) and type II (ZNF9 gene), two other examples of repeat expansion disorders, and that DMPK or ZNF9 transcripts are detected in the foci. Further characterization of the FMR1‐associated inclusions is in progress to address this issue.
Neuronal intranuclear inclusions are well described in brains that are free of evident disease processes. Such non‐pathological inclusions include Marinesco bodies and rodlets of Roncoroni. Marinesco bodies are round, eosinophilic intranuclear inclusions approximately the size of the nucleolus. They are seen in a small percentage of the nuclei of pigmented neurones of the substantia nigra in adults, with some increase in number with advancing age (Yuen and Baxter, 1963). However, Marinesco bodies in neurologically intact individuals have not been observed in areas outside of the substantia nigra and locus coeruleus. Rodlets of Roncoroni have been observed as slender, dark rods within nuclei of neurones in non‐pathological conditions. On ultrastructural analysis, they are composed of non‐membrane‐bound, parallel filamentous arrays (Ghadially, 1997). Both types of non‐pathological inclusion appear to be distinct in morphology, frequency and/or distribution from the inclusions described in the current work.
The presence of diffuse, ubiquitin‐positive cytoplasmic staining in addition to staining of the nuclear inclusions is interesting (Case 1; Fig. 2E), since ubiquitinated proteins generally do not accumulate in healthy cells, but are degraded. While this finding needs confirmation in more cases, similar findings have been reported in Huntington’s disease (Petersen et al., 2001); although we are unaware of any similar findings for other CAG repeat disorders. Using transgenic mice that overexpress the CAG repeat‐containing exon 1 of huntingtin, Petersen et al. (2001) noted that while many cells throughout the body express the abnormal protein product, it is the striatal neurones that undergo cell death. They concluded that dopamine‐induced stress on this group of cells, not the abnormal protein per se, results in formation of ubiquitin‐ and huntingtin‐positive cytoplasmic granules, with some cell lines eventually showing huntingtin‐positive intraneuronal inclusions. Those observations imply that the accumulation of ubiquitinated protein in neurodegenerative disorders may be due to the inability of affected cells to degrade abnormal proteins. This accumulation may be due to an intrinsically lower capacity of the degradation pathway in certain cells, or to increased degradative demands resulting from cellular stress, itself a consequence of altered neuronal function. This ‘altered degradation’ mechanism offers a plausible, general explanation for the presence of inclusions in a variety of neurodegenerative disorders, and the appearance of morphologically similar inclusions in disorders with (e.g. CAG‐repeat disorders) or without (e.g. FXS carriers) abnormal protein products (for a review see Chung et al., 2001). Other neurodegenerative disorders, including Alzheimer and non‐Alzheimer dementias, amyotrophic lateral sclerosis and some transmissible encephalopathies, also show ubiquitin‐positive cytoplasm (Alves‐Rodrigues et al., 1998).
The absence of neuronal intranuclear inclusions in Purkinje cells, as seen in our cases, has also been recognized in the CAG repeat disorders, although one report has noted a few inclusions in Purkinje cells in spinocerebellar ataxia type 7 (Holmberg et al., 1998). Since our cases and most CAG repeat disorders do not show Purkinje cell inclusions, there may be an intrinsic difference in the degradative pathway (or capacity) that precludes the accumulation of ubiquitinated proteins in Purkinje cells. Although the current cases had some Purkinje cell loss, there were sufficient numbers of cells to determine that inclusions were not present at the 1% level. Moreover, in spinocerebellar ataxia type 3/Machado–Joseph disease (Paulson et al., 1997), adult dentatorubral pallidoluysian atrophy (Robitaille et al., 1997) and adult Huntington’s disease (Ross et al., 1997), which also have little Purkinje cell loss, intranuclear inclusions are not observed in that cell type.
The astrocytic intranuclear inclusions were similar in appearance and staining capacity to those seen in neurones. Noteworthy differences were seen in the numbers of inclusions between grey and white matter in both Cases 1 and 2, as well as between the numbers of cortical astrocytic inclusions in the two cases (45 and 14.5%, respectively). Neither the importance of astrocytic inclusions in the disease process nor the variable numbers between the cases is understood at this point.
Clinical–pathological correlates
The FMR1 premutation carriers with neurological findings, previously reported by Hagerman et al. (2001), had progressive cerebellar tremor and ataxia, frontal executive deficits, mild parkinsonian signs and generalized brain atrophy. The current cases had a very similar presentation, except that tremor was a less prominent feature.
In the present study, the degenerative cerebellar findings were present to varied degrees in the three cases in which cerebellar tissue was available for evaluation. The cerebellum, with Purkinje cell loss, dystrophic axons, gliosis and spongy change in cerebellar white matter, was one of the most affected brain regions. These findings correlate with the clinical presentation, since cerebellar ataxia is an early, consistent and major finding in these men. The role of the cerebellum is to coordinate movement, and the Purkinje cells are the only output neurones of the cerebellar cortex. Thus, Purkinje cell loss results in gait ataxia, and probably contributes to the intention tremor present in our previously reported patients (Hagerman et al., 2001). Another important role of the cerebellum is motor learning and cognition. The cerebellum makes a fundamental contribution to executive cognitive functions, such as the planning, reasoning and thinking functions associated with the prefrontal cortex (Hallett and Grafman, 1997). Thus, the cerebellar pathology in these patients probably contributed to their prominent frontal executive deficits as well as their poor motor coordination.
The preponderance of neuronal intranuclear inclusions in the hippocampus, with lesser numbers in cerebral cortex and substantia nigra, closely correlated with the patients’ clinical findings. Prominent hippocampal involvement was consistent with the patients’ memory loss and labile emotion and behaviour. The patients’ intellectual and functional decline also correlated with the widespread cortical involvement. Mild parkinsonian features are consistent with functional abnormalities of the substantia nigra and basal ganglia. One patient (Case 4) had Lewy bodies, which is consistent with co‐occurrence of idiopathic Parkinson’s disease in this individual. The presence of jaw tremor and the levodopa response were clues during life that this individual had idiopathic Parkinson’s disease. His prominent gait instability was not consistent with idiopathic Parkinson’s disease, and probably was secondary to cerebellar involvement related to the FXS premutation. In Cases 1 and 2, the lack of Lewy bodies or dopaminergic cell loss differentiates this syndrome from idiopathic Parkinson’s disease.
Conclusions
Intranuclear inclusions were observed in neuronal and astroglial cells from the brains of four carriers of pre‐mutation alleles of the FMR1 gene. All four males suffered from a newly described neurological condition that involves progressive action tremor and/or gait ataxia as well as cognitive decline. The inclusions are distinct in morphology, frequency of occurrence, distribution and/or immunochemical staining characteristics from previously described pathological and non‐pathological intranuclear inclusions in brain. The inclusions have been found in all four cases examined to date, suggesting that inclusion formation is likely to play a role in the development of the tremor/ataxia disorder among premutation carriers.
Acknowledgements
We wish to thank Brandon Bakhtiar in the UCDMC Optical Biology Lab for cell counts, Grete Adamson of the UCDMC EM Lab for her help with the ultrastructural images, and Dr W. C. Halliday for his input on Case 2. This work was supported by the Boory and Clark family funds and NIH grant GM35305 (P.J.H.), NIH grants HD36071and MO1 RR00069, (R.J.H.), and the UCD MIND Institute (P.J.H., R.J.H., F.T. and C.M.G.).
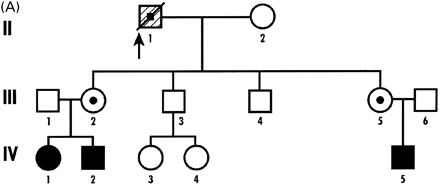
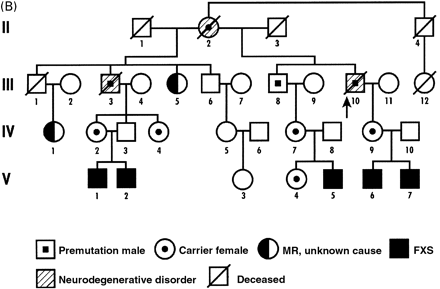
Fig. 1 Pedigree of (A) Case 1 and (B) Case 2.
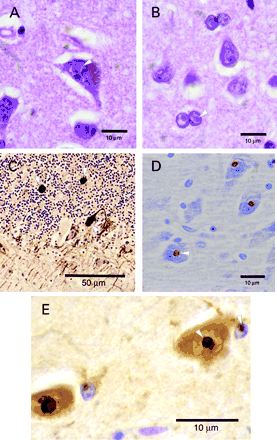
Fig. 2 (A and B) Typical intranuclear inclusions observed in neurones and astroglia, respectively, of Case 1 (H&E stain, original magnification 1000×). (C) Scattered axonal torpedoes in cerebellum, labelled with anti‐neurofilament antibody (original magnification 400×). (D and E) Intranuclear inclusions stained with anti‐ubiquitin antibodies (original magnification 1000×); Cases 2 and 1, respectively. We found non‐specific variability in neuronal cytoplasmic staining by anti‐ubiquitin antibody within individual cases as well as between cases. This variability could have pathological significance, but could also have a methodological and/or technical component.
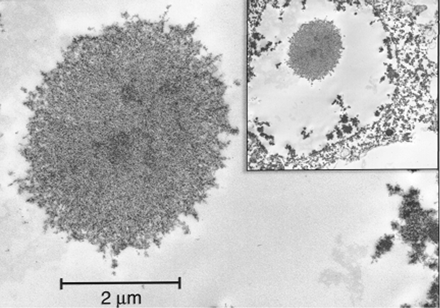
Fig. 3 Electron micrograph of an intranuclear neuronal inclusion (hippocampus, Case 1) consisting of non‐membrane bound, granulofilamentous material (27 950×). Inset: lower magnification view of the inclusion within the nucleus (6500×).
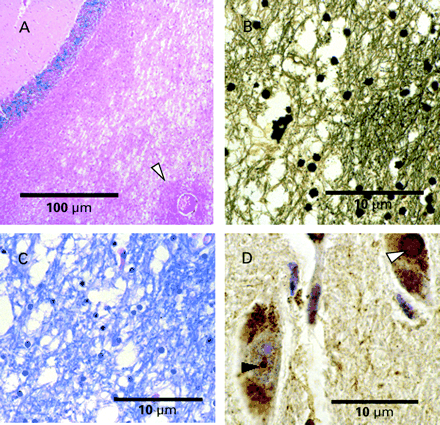
Fig. 4 (A) Marked spongiosis of deep cerebellar white matter is seen in Case 2. The arrowhead indicates relatively normal perivascular white matter (H&E stain, original magnification 100×). (B) Silver stain identifying axonal loss in areas of white matter vacuolation (Bielschowsky stain, original magnification 400×). (C) Myelin stain demonstrating irregular vacuolation and loss of myelin (LFB‐PAS stain, original magnification 400×). (D) Anti‐ubiquitin staining of Case 4 demonstrates both an intracytoplasmic Lewy body (white arrowhead) and an intranuclear inclusion (black arrowhead) within pigmented neurones of the substantia nigra (original magnification 1000×).
Counts of intranuclear inclusion in various brain regions
| Brain area | Case 1 | Case 2 | ||||||
| Neurones | % | Astrocytes | % | Neurones | % | Astrocytes | % | |
| Frontal cortex | 13/237 | 5.5 | 94/209 | 45.0 | 8/233 | 3.4 | 29/200 | 14.5 |
| Temporal cortex | 5/142 | 3.5 | 47/106 | 44.4 | 7/303 | 2.3 | 23/203 | 11.3 |
| Postcentral gyrus | 29/346 | 8.4 | 99/205 | 48.3 | NA | NA | ||
| Calcarine cortex | ND | – | – | – | 4/205 | 2.0 | 33/214 | 10.4 |
| Putamen | 9/213 | 4.2 | 51/113 | 45.1 | 3/70 | 4.3 | 12/162 | 7.4 |
| Globus pallidus | 3/81 | 3.7 | 78/182 | 42.8 | 2/188 | 1.0 | 28/222 | 12.6 |
| Substantia nigra | 63/602 | 10.5 | NC | – | 6/144 | 4.0 | 24/230 | 10.4 |
| Hippocampus | 59/157 | 37.6 | NC | – | 120/279 | 43.0 | NC | |
| Dentate gyrus | 12/422 | 2.8 | NC | – | 5/261 | 1.9 | NC | |
| Basis pontis | 1/185 | 0.5 | 63/112 | 20.2 | 0/200 | 0 | 5/285 | 1.8 |
| Dentate nucleus | 7/210 | 3.3 | 102/208 | 49.0 | 4/150 | 2.7 | 28/168 | 16.7 |
| Purkinje cell layer | 0/150 | 0 | – | |||||
| Brain area | Case 1 | Case 2 | ||||||
| Neurones | % | Astrocytes | % | Neurones | % | Astrocytes | % | |
| Frontal cortex | 13/237 | 5.5 | 94/209 | 45.0 | 8/233 | 3.4 | 29/200 | 14.5 |
| Temporal cortex | 5/142 | 3.5 | 47/106 | 44.4 | 7/303 | 2.3 | 23/203 | 11.3 |
| Postcentral gyrus | 29/346 | 8.4 | 99/205 | 48.3 | NA | NA | ||
| Calcarine cortex | ND | – | – | – | 4/205 | 2.0 | 33/214 | 10.4 |
| Putamen | 9/213 | 4.2 | 51/113 | 45.1 | 3/70 | 4.3 | 12/162 | 7.4 |
| Globus pallidus | 3/81 | 3.7 | 78/182 | 42.8 | 2/188 | 1.0 | 28/222 | 12.6 |
| Substantia nigra | 63/602 | 10.5 | NC | – | 6/144 | 4.0 | 24/230 | 10.4 |
| Hippocampus | 59/157 | 37.6 | NC | – | 120/279 | 43.0 | NC | |
| Dentate gyrus | 12/422 | 2.8 | NC | – | 5/261 | 1.9 | NC | |
| Basis pontis | 1/185 | 0.5 | 63/112 | 20.2 | 0/200 | 0 | 5/285 | 1.8 |
| Dentate nucleus | 7/210 | 3.3 | 102/208 | 49.0 | 4/150 | 2.7 | 28/168 | 16.7 |
| Purkinje cell layer | 0/150 | 0 | – | |||||
NA = not available; NC = inclusions present, not counted; ND = not determined.
Counts of intranuclear inclusion in various brain regions
| Brain area | Case 1 | Case 2 | ||||||
| Neurones | % | Astrocytes | % | Neurones | % | Astrocytes | % | |
| Frontal cortex | 13/237 | 5.5 | 94/209 | 45.0 | 8/233 | 3.4 | 29/200 | 14.5 |
| Temporal cortex | 5/142 | 3.5 | 47/106 | 44.4 | 7/303 | 2.3 | 23/203 | 11.3 |
| Postcentral gyrus | 29/346 | 8.4 | 99/205 | 48.3 | NA | NA | ||
| Calcarine cortex | ND | – | – | – | 4/205 | 2.0 | 33/214 | 10.4 |
| Putamen | 9/213 | 4.2 | 51/113 | 45.1 | 3/70 | 4.3 | 12/162 | 7.4 |
| Globus pallidus | 3/81 | 3.7 | 78/182 | 42.8 | 2/188 | 1.0 | 28/222 | 12.6 |
| Substantia nigra | 63/602 | 10.5 | NC | – | 6/144 | 4.0 | 24/230 | 10.4 |
| Hippocampus | 59/157 | 37.6 | NC | – | 120/279 | 43.0 | NC | |
| Dentate gyrus | 12/422 | 2.8 | NC | – | 5/261 | 1.9 | NC | |
| Basis pontis | 1/185 | 0.5 | 63/112 | 20.2 | 0/200 | 0 | 5/285 | 1.8 |
| Dentate nucleus | 7/210 | 3.3 | 102/208 | 49.0 | 4/150 | 2.7 | 28/168 | 16.7 |
| Purkinje cell layer | 0/150 | 0 | – | |||||
| Brain area | Case 1 | Case 2 | ||||||
| Neurones | % | Astrocytes | % | Neurones | % | Astrocytes | % | |
| Frontal cortex | 13/237 | 5.5 | 94/209 | 45.0 | 8/233 | 3.4 | 29/200 | 14.5 |
| Temporal cortex | 5/142 | 3.5 | 47/106 | 44.4 | 7/303 | 2.3 | 23/203 | 11.3 |
| Postcentral gyrus | 29/346 | 8.4 | 99/205 | 48.3 | NA | NA | ||
| Calcarine cortex | ND | – | – | – | 4/205 | 2.0 | 33/214 | 10.4 |
| Putamen | 9/213 | 4.2 | 51/113 | 45.1 | 3/70 | 4.3 | 12/162 | 7.4 |
| Globus pallidus | 3/81 | 3.7 | 78/182 | 42.8 | 2/188 | 1.0 | 28/222 | 12.6 |
| Substantia nigra | 63/602 | 10.5 | NC | – | 6/144 | 4.0 | 24/230 | 10.4 |
| Hippocampus | 59/157 | 37.6 | NC | – | 120/279 | 43.0 | NC | |
| Dentate gyrus | 12/422 | 2.8 | NC | – | 5/261 | 1.9 | NC | |
| Basis pontis | 1/185 | 0.5 | 63/112 | 20.2 | 0/200 | 0 | 5/285 | 1.8 |
| Dentate nucleus | 7/210 | 3.3 | 102/208 | 49.0 | 4/150 | 2.7 | 28/168 | 16.7 |
| Purkinje cell layer | 0/150 | 0 | – | |||||
NA = not available; NC = inclusions present, not counted; ND = not determined.
References
Allingham‐Hawkins DJ, Babul‐Hirji R, Chitayat D, Holden JJ, Yang KT, Lee C, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study—preliminary data.
Alves‐Rodrigues A, Gregori L, Figueiredo‐Pereira ME. Ubiquitin, cellular inclusions and their role in neurodegeneration. [Review].
Brown WT, Houck GE, Jeziorowska A, Levinson FN, Ding X, Dobkin C, et al. Rapid fragile X carrier screening and prenatal diagnosis using a nonradioactive PCR test.
Chung KK, Dawson VL, Dawson TM. The role of the ubiquitin–proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders.
Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR‐1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation.
Dorn MB, Mazzocco MM, Hagerman RJ. Behavioral and psychiatric disorders in adult male carriers of fragile X.
Evert BO, Wüllner U, Klockgether T. Cell death in polyglutamine diseases. [Review].
Franke P, Leboyer M, Gansicke M, Weiffenbach O, Biancalana V, Cornillet‐Lefebre P, et al. Genotype–phenotype relationship in female carriers of the premutation and full mutation of FMR‐1.
Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox.
Ghadially FN. Ultrastructural pathology of the cell and matrix. 4th edn. Boston: Butterworth‐Heinemann;
Hagerman RJ, Staley LW, O’Connor R, Lugenbeel K, Nelson D, McLean SD, et al. Learning‐disabled males with a fragile X CGG expansion in the upper premutation size range.
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X.
Hallett M, Grafman J. Executive function and motor skill learning. [Review].
Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome.
Holmberg M, Duyckaerts C, Dürr A, Cancel G, Gourfinkel‐An I, Damier P, et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions.
Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile‐X mental retardation syndrome. [Review].
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile‐X syndrome: a quantitative examination.
Kakita A, Oyanagi K, Nagai H, Takahashi H. Eosinophilic intranuclear inclusions in the hippocampal pyramidal neurons of a patient with amyotrophic lateral sclerosis.
Kirkilionis AJ, Chudley AE, Greenberg CR, Yan DL, McGillivray B, Hamerton JL. Transmission of the fra(X) haplotype from three nonpenetrant brothers to their affected grandsons.
Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9.
Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization.
Loesch DZ, Hay DA, Mulley J. Transmitting males and carrier females in fragile X—revisited.
Malandrini A, Villanova M, Tripodi S, Palmeri S, Sicurelli F, Parrotta E, et al. Neuronal intranuclear inclusion disease: neuropathologic study of a case.
Mogk RL, Carson NL, Chudley AE, Dawson AJ. Transmission of the FRAXA haplotype from three nonpenetrant brothers to their affected grandsons: an update with AGG interspersion analysis.
Mostofsky SH, Mazzocco MM, Aakalu G, Warsofsky IS, Denckla MB, Reiss AL. Decreased cerebellar posterior vermis size in fragile X syndrome: correlation with neurocognitive performance.
Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, et al. Instability of a 550‐base pair DNA segment and abnormal methylation in fragile X syndrome.
Paulson HL, Das SS, Crino PB, Perez MK, Patel SC, Gotsdiner D, et al. Machado–Joseph disease gene product is a cytoplasmic protein widely expressed in brain.
Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, et al. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine‐mediated striatal neuron autophagy and degeneration.
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, et al. Absence of expression of the FMR‐1 gene in fragile X syndrome.
Reiss AL, Aylward E, Freund LS, Joshi PK, Bryan RN. Neuroanatomy of fragile X syndrome: the posterior fossa.
Reiss AL, Freund L, Tseng JE, Joshi PK. Neuroanatomy in fragile X females: the posterior fossa.
Reiss AL, Lee J, Freund L. Neuroanatomy of fragile X syndrome: the temporal lobe. [Review].
Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB. Neurodevelopmental effects of the FMR1 full mutation in humans.
Reyniers E, Martin JJ, Cras P, Van Marck E, Handig I, Jorens HZ, et al. Postmortem examination of two fragile X brothers with an FMR1 full mutation.
Riddle JE, Cheema A, Sobesky WE, Gardner SC, Taylor AK, Pennington BF, et al. Phenotypic involvement in females with the FMR1 gene mutation.
Robitaille Y, Lopes‐Cendes I, Becher M, Rouleau G, Clark AW. The neuropathology of CAG repeat diseases: review and update of genetic and molecular features. [Review].
Ross CA, Becher MW, Colomer V, Engelender S, Wood JD, Sharp AH. Huntington’s disease and dentatorubral‐pallidoluysian atrophy: proteins, pathogenesis and pathology. [Review].
Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K. Prevalence of carriers of premutation‐size alleles of the FMRI gene—and implications for the population genetics of the fragile X syndrome.
Rousseau F, Morel M‐L, Rouillard P, Khandjian EW, Morgan K. Surprisingly low prevalance of FMR1 premutations among males from the general population [abstract].
Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure‐Kamionowska M, Connell F, et al. Adult fragile X syndrome. Clinico‐neuropathologic findings.
Sabaratnam M. Pathological and neuropathological findings in two males with fragile‐X syndrome. [Review].
Schapiro MB, Murphy DG, Hagerman RJ, Azari NP, Alexander GE, Miezejeski CM, et al. Adult fragile X syndrome: neuropsychology, brain anatomy, and metabolism.
Smits A, Smeets D, Hamel B, Dreesen J, de Haan A, van Oost B. Prediction of mental status in carriers of the fragile X mutation using CGG repeat length.
Snow K, Doud LK, Hagerman R, Pergolizzi RG, Erster SH, Thibodeau SN. Analysis of a CGG sequence at the FMR‐1 locus in fragile X families and in the general population.
Takahashi J, Fukuda T, Tanaka J, Minamitani M, Fujigasaki H, Uchihara T. Neuronal intranuclear hyaline inclusion disease with polyglutamine‐immunoreactive inclusions.
Tassone F, Hagerman RJ, Iklé DN, Dyer PN, Lampe M, Willemsen R, et al. FMRP expression as a potential prognostic indicator in fragile X syndrome.
Tassone F, Hagerman RJ, Chamberlain WD, Hagerman PJ. Transcription of the FMR1 gene in individuals with fragile X syndrome.
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile‐X syndrome.
Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW, et al. Clinical involvement and protein expression in individuals with the FMR1 premutation.
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR‐1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome.
Willemsen R, Mohkamsing S, de Vries B, Devys D, van den Ouweland A, Mandel JL, et al. Rapid antibody test for fragile X syndrome.
Willemsen R, Smits A, Mohkamsing S, van Beerendonk H, de Haan A, de Vries B, et al. Rapid antibody test for diagnosing fragile X syndrome: a validation of the technique.
Yamada M, Tsuji S, Takahashi H. Pathology of CAG repeat diseases. [Review].
Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, et al. Fragile X genotype characterized by an unstable region of DNA.
Author notes
1Department of Pathology, 2MIND Institute and 3Department of Biological Chemistry, University of California, Davis, School of Medicine, Davis, CA, 4Department of Neurology, University of Colorado Health Sciences Center, Denver, CO, USA, 5Department of Pediatrics and Child Health, University of Manitoba, and Section of Genetics and Metabolism, Children’s Hospital, 6Department of Pathology, University of Manitoba, Winnipeg, Manitoba, Canada


{kind=link}
{kind=link}
{kind=link}
{kind=link}