




Abstract
Patients presenting with subacute amnesia are frequently seen in acute neurological practice. Amongst the differential diagnoses, herpes simplex encephalitis, Korsakoff’s syndrome and limbic encephalitis should be considered. Limbic encephalitis is typically a paraneoplastic syndrome with a poor prognosis; thus, identifying those patients with potentially reversible symptoms is important. Voltage‐gated potassium channel antibodies (VGKC‐Ab) have recently been reported in three cases of reversible limbic encephalitis. Here we review the clinical, immunological and neuropsychological features of 10 patients (nine male, one female; age range 44–79 years), eight of whom were identified in two centres over a period of 15 months. The patients presented with 1–52 week histories of memory loss, confusion and seizures. Low plasma sodium concentrations, initially resistant to treatment, were present in eight out of 10. Brain MRI at onset showed signal change in the medial temporal lobes in eight out of 10 cases. Paraneoplastic antibodies were negative, but VGKC‐Ab ranged from 450 to 5128 pM (neurological and healthy controls <100 pM). CSF oligoclonal bands were found in only one, but bands matched with those in the serum were found in six other patients. VGKC‐Abs in the CSF, tested in five individuals, varied between <1 and 10% of serum values. Only one patient had neuromyotonia, which was excluded by electromyography in seven of the others. Formal neuropsychology testing showed severe and global impairment of memory, with sparing of general intellect in all but two patients, and of nominal functions in all but one. Variable regimes of steroids, plasma exchange and intravenous immunoglobulin were associated with variable falls in serum VGKC‐Abs, to values between 2 and 88% of the initial values, together with marked improvement of neuropsychological functioning in six patients, slight improvement in three and none in one. The improvement in neuropsychological functioning in seven patients correlated broadly with the fall in antibodies. However, varying degrees of cerebral atrophy and residual cognitive impairment were common. Over the same period, only one paraneoplastic case of limbic encephalitis was identified between the two main centres. Thus, VGKC‐Ab‐associated encephalopathy is a relatively common form of autoimmune, non‐paraneoplastic, potentially treatable encephalitis that can be diagnosed by a serological test. Establishing the frequency of this new syndrome, the full range of clinical presentations and means of early recognition, and optimal immunotherapy, should now be the aim.
Introduction
The term limbic encephalitis was originally coined by Corsellis et al. (1968), and refers to the subacute onset of episodic memory impairment, disorientation and agitation (Brierley et al., 1960), commonly associated with seizures, hallucinations, sleep disturbance and histological evidence of medial temporal lobe inflammation. Signal changes in the medial temporal lobes or hippocampi are frequently found on MRI. Limbic encephalitis is usually considered to be paraneoplastic in origin, and many reported cases are associated with specific autoantibodies; mainly to Hu in patients with lung cancer (Dalmau et al., 1992; Alamowitch et al., 1997; Graus et al., 2001), to Ma2 in patients with testicular tumours (Voltz et al., 1999) or to CRMP5/CV2 in patients with thymomas (Antoine et al., 1995). In those cases with thymoma, with or without myasthenia gravis (Antoine et al., 1995), or in cases with Ma2 antibodies (Gultekin et al., 2000; Rosenfeld et al., 2001), there may be improvement after treatment of the primary tumour, but in general the prognosis of paraneoplastic limbic encephalitis is poor. A few cases of non‐paraneoplastic limbic encephalitis have been described (Bien et al., 2000; Mori et al., 2002), but their immunological basis was not established.
In contrast, limbic encephalitis associated with voltage‐gated potassium channel antibodies (VGKC‐Abs) (Buckley et al., 2001; Schott et al., 2003), may frequently be non‐paraneoplastic. In two of the three patients described, there was a marked and sustained improvement following immunosuppressive therapy, whilst the other improved spontaneously in parallel with a fall in VGKC‐Abs to near normal levels. A recent study of 15 cases of limbic encephalitis found raised VGKC‐Abs in four, with the two highest levels (>400 pM) associated with non‐paraneoplastic disorders and remission following immunosuppressive treatment (Pozo‐Rosich et al., 2003). VGKC‐Abs have previously been implicated in acquired neuromyotonia and related disorders involving the peripheral nervous system (Shillito et al., 1995; Hart et al., 1997, 2002; Vernino and Lennon, 2002), but they are also found in Morvan’s syndrome, a rare condition in which neuromyotonia is accompanied by autonomic disturbance, sleep and cognitive disorders (Lee et al., 1998; Barber et al., 2000; Liguori et al., 2001).
These observations led us to look for VGKC‐Abs in patients presenting with subacute amnestic syndromes of unknown cause. Here we describe the clinical features, and serological and neuropsychological changes in 10 patients who presented within a 15‐month period to Oxford (five), London (three), Halifax (one) or Bonn (one). All had clinical features compatible with a subacute encephalopathy, no evidence of occult malignancy and highly elevated serum titres of VGKC‐Abs. Six of them have improved substantially following immunotherapy, associated with dramatic and sustained falls in serum VGKC‐Ab titres.
Patients and methods
Patients
We included patients who presented with clinical features of a subacute amnesic encephalopathy compatible with a diagnosis of paraneoplastic limbic encephalitis but who had no evidence of a tumour, negative results for paraneoplastic antibodies and highly raised VGKC‐Abs (>400 pM). Control samples were taken from consecutive patients attending one of the clinical centres and gave values of <100 pM (Fig. 1A). We did not include patients with VGKC‐Abs between 100 and 400 pM because we have found these values relatively frequently (5%) in a study of 164 elderly subjects (whose neurological status is unknown) attending non‐neurological clinics at the John Radcliffe Hospital in Oxford. The clinical courses, investigations and responses to treatment were reviewed retrospectively from the case notes. CSF studies, MRI and EEG were performed in all patients, and EMG in most.
Neuropsychology
Since the study was retrospective, the patients were not investigated in a standard manner. The following domains were evaluated where data was available. Pretreatment level of functioning was assessed with the National Adult Reading Test—Revised (NART‐R; Nelson, 1991) in five patients. The pretreatment functioning of Case 2 was measured with the Schonell Graded Reading Test (Nelson et al., 1975). Current intellectual functioning was assessed with the Wechsler Adult Intelligence Scale—Revised (WAIS‐R; Wechsler, 1981) or WAIS‐III (Wechsler, 1997). A variety of tasks was used to assess memory function across patients: the Recognition Memory Test (RMT; Warrington, 1984), story recall and list learning tasks from the Adult Memory and Information Processing Battery (Coughlan and Hollows, 1985) and the Rey Complex Figure Test (Osterreith, 1944; Rey, 1964). Nominal skills were assessed with the Graded Naming Test (McKenna and Warrington, 1980). The pretreatment and current intellectual functioning scores are represented as IQ scores. A difference between the NART and IQ of ≥10 was taken as evidence of intellectual decline. The memory and naming scores were derived by converting the standardized test performance into percentile scores. Scores at or below the 5th percentile were taken to indicate impairment.
Serology
All patients’ sera were negative for paraneoplastic antibodies on routine screening. In some cases, the presence of VGKC‐Abs was suspected during this screen (see Results). Serum VGKC‐Ab titres were measured by radioimmunoassay using whole rabbit‐brain homogenate as described previously (Shillito et al., 1995; Hart et al., 1997; L. Clover et al., in preparation). This assay measures antibodies to the VGKC subtypes, Kv1.1, 1.2 and 1.6, that bind 125I‐labelled dendrotoxin. These subtypes are expressed in both the CNS and PNS. Sera were first screened using 5 µl of serum; sera immunoprecipitating >400 pmol of dendrotoxin binding sites per litre (400 pM) were retested using 0.3125–2.5 µl serum to obtain accurate values. All sera were also tested for binding to rat brain by immunohistochemistry with a 1:200 dilution of serum on acetone‐fixed frozen sections of cerebellum and brainstem, as described elsewhere (Amyes et al., 2001).
Results
The case reports below describe the histories of three of the patients up to the end of June 2003. The remaining case histories are available from Brain online.
Case 1
This 57‐year‐old male was admitted to hospital in July 2002 with a 36‐h history of amnesia and disorientation. He had been made redundant from his job as a facilities manager for a telephone company, and around this time suffered from depression, newly diagnosed diabetes mellitus and weight loss. There was a past history of pernicious anaemia and a family history of systemic lupus erythematosus. On admission, excess sweating and salivation were noted. Initial neuropsychological assessment indicated normal intellectual ability, but impaired verbal and visual memory. There was also a 5‐year retrograde amnesia, and confabulation. Neuromyotonia was present both on clinical and neurophysiological examination. There was hyponatraemia due to syndrome of inappropriate secretion of antidiuretic hormone (SIADH), and raised C‐reactive protein and gamma‐glutaryltranspeptidase levels. The patient subsequently suffered a grand mal seizure. T2‐weighted MRI of the brain demonstrated bilateral hippocampal high signal (Fig. 2A) but the EEG was normal. Paraneoplastic antibodies, CT chest and vasculitis screen were normal or negative. Serum VGKC‐Abs were 3120 pM. CSF examination showed a white cell count of 11 and a mildly elevated protein level of 0.75 g/l. Polymerase chain reaction (PCR) for herpes virus was neagative. Oligoclonal bands were present, matched in serum and CSF. VGKC‐Abs were present in the CSF at 350 pM, representing 11% of the matched serum value. Although there was no history of excess alcohol intake, treatment with parenteral thiamine was given. In view of the raised VGKC‐Ab titre, the patient underwent 5 days of plasma exchange and was started on 100 mg alternate day oral prednisolone. Because there was no evident improvement after 3 weeks, a standard course of intravenous immunoglobulin (IvIg; 2g/kg divided into five daily doses) was given. Improvement began shortly after: insight returned, confabulation ceased and the patient became fully orientated. On repeat neuropsychological testing 3 months later, there was a dramatic improvement in verbal and non‐verbal memory, although a selective impairment in delayed verbal recall remained. A second MRI performed 6 months after the first demonstrated resolution of the previous hippocampal T2 high signal, but now revealed bilateral hippocampal atrophy. The patient continues treatment with corticosteroids under active follow‐up, and VGKC‐Abs are currently <100 pM, representing a fall of >98% of the value at onset (see Fig. 1B).
Case 4
This 71‐year‐old retired telephone engineer initially presented to his general practitioner complaining of occasional confusion following a flu‐like illness. Over a 2‐month period these symptoms worsened, he lost >5 kg in weight and his wife described episodes of absence and possible automatisms. On admission to hospital, the Mini‐Mental State Examination (MMSE) score was 16/30 and there was profound memory impairment, but physical examination was normal. The plasma sodium was found to be 130 mmol/l, due to SIADH, and he was placed on fluid restriction. Despite this, 12 days later his sodium dropped to 113 mmol/l and he suffered two tonic–clonic seizures. Further investigations revealed a raised C‐reactive protein level and abnormal liver function tests. Neuropsychological testing revealed a global deterioration of intellectual functioning and a severe impairment of verbal and visual memory. EEG showed slow waves and focal sharp waves over the right frontal and anterior temporal regions, and an MRI brain scan showed cerebral atrophy, with signal change and selective enlargement of the left hippocampus, and subtle subcortical high signal in the right insular region, consistent with encephalitis. CSF examination was unremarkable, but the serum VGKC‐Ab titre was elevated at 1232 pM. Treatment commenced with a 5‐day course of IvIg and 100 mg alternate day prednisolone. Improvement was noted within the first week and repeat neuropsychological testing at 3 months confirmed a return of general intellectual ability to estimated pretreatment levels, decreased confusion and improved memory function. However, there was still an ongoing selective impairment in delayed verbal recall. After 6 months of oral corticosteroids, by which time the VGKC‐Abs had fallen by 89% to 131 pM (Fig. 1B), his MRI had normalized and his family reported no obvious day to day memory problems, although he had become more placid and had not regained his drive. The corticosteroid dose is currently being reduced.
Case 8
This 64‐year‐old taxi driver presented with a 6‐week history of rapidly progressive amnesia and confusion, 2 weeks after a diarrhoeal illness. His wife reported that he had become apathetic and lost empathy. His memory was impaired as he began to ask repetitive questions and required the use of a map to continue his work. His MMSE score was 13/30, and he was disorientated for time and place. He was treated with intravenous aciclovir. Sodium levels were initially normal (137 mmol/l), but decreased to a nadir of 125 mmol/l, attributed to SIADH. A CSF examination revealed matched oligoclonal bands, but was otherwise unremarkable; PCR for herpes viruses was negative. Paraneoplastic autoantibodies, thyroid antibodies, tumour markers and infectious serology were negative or normal, as were EMG, chest CT and whole‐body FDG‐PET ([18F]fluorodeoxyglucose–PET). Antinuclear antibodies were mildly elevated at 1/320. EEG showed generalized mild background slowing. Brain MRI revealed increased signal in both hippocampi and anterior temporal lobes (Fig. 2C). VGKC‐Abs were found, retrospectively, to be 709 pM. Neuropsychological testing revealed global memory impairment and a dysexecutive syndrome. Four months later, he developed complex partial and tonic–clonic seizures. At that time the sodium was 125 mmol/l. Oligoclonal bands were no longer present in the CSF. The EEG now revealed diffuse slowing with a paucity of alpha rhythm but the MRI showed a decline in medial temporal lobe signal change. He was treated with phenytoin, which produced a rash, necessitating a change to sodium valproate and clobazam. He received a 5‐day course of IvIg. His seizures increased in frequency, requiring admission to intensive care where he received phenobarbitone and lorazepam, and then underwent plasma exchange. The VGKC‐Ab level had risen to 1132 pM. His condition slowly improved, although he developed a leucocytoclastic rash, thought to represent a drug reaction to sodium valproate or antibiotics. Repeat neuropsychological assessment indicated some improvement of verbal memory, and his VGKC‐Abs had fallen to 803 pM. He was discharged to a regional brain injury unit, where he remained disoriented, with very poor memory and recognition. At follow‐up 4 months later, his condition had stabilized, with no further improvement in cognitive state, and occasional blank spells, thought to represent seizures. The sodium level was now normal, and the MRI showed mild atrophy with some minimal signal change in the hippocampi; the VGKC‐Ab levels were still substantially elevated at 665 pM (Fig. 1C). The patient was latterly started on high dose oral corticosteroids.
Clinical features
The patients were defined by the presence of limbic encephalitis and strongly positive VGKC‐Abs. Case 2 has been reported previously (Schott et al., 2003). The clinical details of Cases 2, 3, 5–7, 9 and 10 are available at Brain online, and the main features of all patients are summarized in Table 1. Several patients were not seen by the relevant author until weeks or months following the initial symptoms. The median age was 57 years (range 44–79 years); there were nine males and one female. Eight presented with combinations of impaired episodic memory, confusion and disorientation, and all patients developed these symptoms early during their illness. Seizures were present in nine patients during the acute phase of the disease, including grand mal and/or complex partial seizures. Additional features included hallucinations, agitation and behavioural disturbance. Two patients had gait disturbance, but the neurological abnormalities could all be attributed to limbic dysfunction in the others. Headache, drowsiness and loss of consciousness were not present in general, although Case 2 had a prolonged period of obtundation (see Schott et al., 2003).
One patient had a history of late onset depression. There was a history of prodromal illness, presumed to be viral infection, in four patients, but no other common features leading up to the onset of the disease. Only Case 1 complained of muscle twitching or cramps and he also had excessive sweating. Myoclonus was noticed in three others but was probably central, rather than peripheral, in origin, since routine EMG was normal, as it was in the three cases without muscle symptoms (see below). Two of the patients had an adverse reaction to phenytoin.
Clinical investigations
Table 2 summarizes the relevant clinical investigations. A striking feature was the low plasma sodium concentrations in eight of the 10 cases, which were either present on admission or developed subsequently. Several were confirmed to be SIADH, which was often difficult to treat, although it eventually resolved completely in all cases. Lumbar puncture revealed a mild lymphocytosis in five cases; the protein and glucose were modestly raised or within normal limits. PCR was negative for herpes simplex virus in all patients tested. Oligoclonal bands were present in the CSF in five cases, but in four of these there were matched serum bands, and the bands had disappeared on a second sampling in those tested. MRI showed bilateral medial temporal lobe high signal in five of the patients, either at presentation or within 2 weeks, left‐sided hippocampal high signal in three and no abnormality in two. Electroencephalography usually showed non‐specific changes with generalized slowing, with focal sharp waves especially in the temporal regions in some cases; Case 10 had left‐sided temporal slowing. Case 10 had a stereotactic biopsy of the left amygdala that demonstrated perivascular and parenchymal lymphocytes, astrogliosis and microglial activity (Fig. 3). EMG showed neuromyotonic discharges in Case 1, and no abnormalities in the other seven tested. A paraneoplastic origin for their symptoms was considered carefully in each patient, but tumour markers and CT, performed if appropriate, were negative.
Immunological investigations
Patient 8 had anti‐nuclear antibodies at low titre. Case 10 had reduced thyroid stimulating hormone levels, which normalized spontaneously in the absence of thyroid autoantibodies. None of the sera were positive for paraneoplastic antibodies. However, two of the six patients who were first tested for paraneoplastic antibodies in Oxford showed binding to the molecular layer of the rat cerebellum sparing the Purkinje cells during routine testing (e.g. Case 3, Fig. 4). This pattern of antibody binding is distinct from that of any of the known paraneoplastic antibodies, but has previously been recognized in sera from some patients with Morvan’s syndrome who have high VGKC‐Abs (E. Tüzun and A. Vincent, unpublished observations). The 10 sera were therefore retested for binding to rat brain sections, and the five that were strongly positive for VGKC‐Abs (>2000 pM) by radioimmunoprecipitation were also positive by immunohistochemistry.
In five patients it was possible to perform VGKC‐Ab assays on matched serum and CSF (Table 2). CSF antibodies were present in four, at levels between 1 and 10% of the serum values, but <10 pM in the CSF from Case 6 who had the lowest serum values (450 pM).
Neuropsychological testing
Owing to the retrospective nature of the current study, the patients were not investigated in a standard manner. Seven patients underwent neuropsychological assessment before and after treatment (see Table 3A). Before treatment, there was evidence of general intellectual decline in two patients, Cases 2 and 4 (see Table 3AA), and mild intellectual impairment in Case 9. All patients, with two exceptions (Cases 6 and 9), showed a severe and global impairment of verbal and visual memory. Case 6 presented with spared visual memory and adequate performance on one out of two verbal memory tests. Case 9 presented with a relative sparing of verbal recognition, immediate verbal and visual recall memory.
Clinical, serological and neuropsychological responses to treatment
A summary of the treatments and the clinical responses are shown in Table 4. All but one of the patients had either a standard 5‐day course of plasma exchange (two individuals), IvIg (0.4 mg/kg/day; two individuals) or both (five individuals), followed in most cases by a course of oral corticosteroids (seven individuals). Overall, six out of 10 were judged to have sustained definite, three out of 10 slight and one out of 10 no clinical improvement, ranging from resolution of SIADH (eight out of eight), cessation (eight out of nine) or reduction (one out of nine) of seizure activity, and improvement in memory. Treatment was associated with dramatic falls in VGKC‐Abs to between 2 and 16% of the initial levels in Cases 1–5 (Fig. 1A), with evident clinical improvement including a reduction in seizure frequency. The VGKC‐Abs in the Cases 6–10 (Fig. 1C) showed slower falls to between 17 and 88%, and their clinical improvement was more variable. Seven had follow‐up MRI scans that showed partial or full resolution of the imaging abnormalities consistent with improvement in clinical symptoms, although five showed marked atrophy. All patients, except for Case 3, who died of pneumonia 3 months after diagnosis, are well and continue to improve. There was no evidence of cancer in any patient at time of submission.
The post‐treatment neuropsychological tests are shown in Table 3BB, along with the percentage fall in VGKC‐Abs at the time of the repeat assessments, which took place at variable time intervals ranging from 2 weeks to 12 months later. Cases 2 and 4 showed significant improvement in intellectual functioning following treatment, to a level commensurate with estimated pretreatment abilities. Cases 1 and 4 had improved substantially apart from ongoing poor performance on a verbal list‐learning task. In two patients (Cases 2 and 8), verbal memory function improved selectively, and their visual memory function remained impaired. Some improvements in memory function were noted for all patients, although this was variable and not marked in Cases 6–8. Case 6 showed a mixed response with relative improvement on story recall, but deterioration on list learning, probably reflecting continuing attention/executive dysfunction. Case 7’s verbal and visual recall memory remained impaired. For one patient (Case 9), whose memory was relatively spared at the first testing, memory performance was in the normal range following treatment. Overall, despite the retrospective nature of the study and different treatment regimens used, there was an association between fall in VGKC‐Ab levels and improvement in memory percentile scores, which just failed to reach significance (P = 0.058; Fig. 5).
Discussion
We describe 10 patients with an immunotherapy‐responsive form of limbic encephalitis strongly associated with VGKC‐Abs. The patients presented with a syndrome indistinguishable from the encephalopathies seen with herpes simplex encephalitis, Korsakoff’s syndrome or paraneoplastic limbic encephalitis, with memory loss, confusion and disorientation at presentation, and seizures developing at some time during the acute phase of the illness. The predominant feature of initial neuropsychological investigation was a pervasive and generalized impairment of memory. Although follow‐up was too short formally to exclude malignancies, particularly in Case 3, who died within 3 months of onset, there was, and continues to be, no evidence for neoplasia in any of these cases and none of their sera were positive for the antibodies that are typically associated with paraneoplastic limbic encephalitis. In contrast, all the patients had highly raised VGKC‐Abs when first studied and the majority showed clinical and neuropsychological improvement, correlating broadly with reductions in antibody levels, following immunosuppressive therapy. Persistent symptoms, predominantly amnesia, seen in some of the cases, may be the result of persistent VGKC‐Abs and/or cerebral (and particularly medial temporal lobe) atrophy. Although the majority were male in this study, the condition has also been identified in a number of females (Buckley et al., 2001; A. Vincent, unpublished observations).
Our patients presented with a neuropsychological profile characterized by marked and generalized memory impairment, which is one of the hallmarks of limbic encephalitis (Corsellis, 1968; Lishman, 1998). Mild intellectual impairment was seen in only two patients and nominal dysfunction was evident only in one. Following treatment, memory function improved for all patients, with the exception of Case 7. Recovery was not uniform and ranged from a return to normal memory performance to ongoing selective impairments in verbal memory or visual memory. This suggests that VGKC‐Ab‐associated limbic encephalitis is, at least in part, a potentially reversible amnesic syndrome. Recovery of neuropsychological function following treatment has previously been reported in a case of paraneoplastic limbic encephalitis (Bak et al., 2001). These authors suggested that cognitive deficits extending beyond anterograde amnesia and evidence of destructive medial temporal lobe pathology on imaging were poor prognostic features of paraneoplastic limbic encephalitis; nevertheless, in this study several patients with cerebral atrophy demonstrated a good functional recovery. Future prospective studies systematically assessing a wide range of cognitive domains, including frontal executive function, may help to elucidate prognostic features of this disease.
In this retrospective study, treatments were varied, and improvement following treatments was sometimes delayed. It is not yet clear when maximal improvement occurs; Case 1, for instance, is still showing improvement 1 year after treatment at a time when his VGKC‐Abs are within normal limits. In general, improvements in memory function appear to have been greater in those patients treated with prolonged courses of oral steroids. Importantly, within the time frame of this study, there was little evidence of spontaneous improvement (compare with Buckley et al., 2001).
Our results strongly support a pathogenic role for the VGKC‐Abs in these limbic encephalitis cases. First, there was a striking temporal relationship between clinical improvement and reduction in antibody titre in several patients, and the VGKC‐Abs were present in CSF as well as sera, although at reduced levels consistent with extrathecal synthesis (Table 2). Conversely, there was less or slower reduction in VGKC‐Ab titres in those patients whose improvement was less clear‐cut (Fig. 1C). Secondly, we have previously shown strong immunostaining of the molecular layer of the dentate gyrus with serum antibodies from a patient with VGKC‐Abs and limbic encephalitis (Buckley et al., 2001), showing that the hippocampus is a major potential site of action of these antibodies. The antibodies detected by the radioimmunoprecipitation assay in use here are directed mainly against Kv 1.1 and Kv 1.2 subtypes of VGKCs that are present in the molecular layer of the dentate gyrus (Rhodes et al., 1996; Monaghan et al., 2001). Finally, point mutations in the human Kv 1.1 gene can cause episodic ataxia, myokymia and epilepsy in some patients (Zuberi et al., 1999), and Kv1.1 knockout mice display frequent spontaneous seizures in adult life that are thought to be of limbic origin (Smart et al., 1998), and can also suffer from memory problems (Gratacos et al., 1998).
Many of the antibodies associated with neurological conditions, and particularly with paraneoplastic limbic encephalitis, are directed against intracellular targets (Dalmau et al., 1999; Vincent, 2002), and hence their pathological importance remains undetermined. In contrast, as we have discussed previously (Buckley et al., 2001), VGKCs located on the plasma membrane are key determinants of neuronal excitability, and are known to be the targets for pathogenic antibodies in acquired neuromyotonia (Vernino and Lennon, 2002; Hart et al., 2002). The absence of neuromyotonia in most of the patients studied here is therefore interesting and remains unexplained. Myoclonus was present in three patients, but was probably central in origin, as routine EMGs were normal. Although the assay detects VGKC‐Abs in patients with both neuromyotonia and limbic encephalitis, it may be that the principle antigenic target differs between the two patient groups. The predominance of peripheral or central symptoms could be determined either by differences in antibody affinity and specificity for the different subunits, or by differences in subunit composition of the VGKC channels in the peripheral nerves or limbic structures in different patients. How the antibodies enter the CNS and why they cause mainly limbic symptoms is not clear. Local changes in permeability of the blood–brain barrier, perhaps combined with the sensitivity of neuronal function to loss of VGKCs, may determine where the antibodies gain access to the CNS, and whether they cross in sufficient amounts to cause symptoms. Experimental studies in animal models are required to determine whether VGKC‐Abs alone can cause seizures and memory loss, and how the antibodies get into the CNS.
Hyponatraemia, due to SIADH or cerebral salt wasting, is well recognized to occur following a wide range of cerebral insults including head injury and meningitis (Palmer, 2003). Hyponatraemia was found in the majority of our patients and was characterized by its initial resistance to treatment; it preceded anticonvulsants and other candidate treatments ruling out iatrogenic causes. Additionally, it resolved as the VGKC‐Abs declined. Antibody‐induced reduction in VGKCs in the motor nerve terminal is thought to lead to increased neurotransmitter release (Shillito et al., 1995). It is therefore possible that the sweating and hypersecretion seen in Case 1 (and in our previous report, see Buckley et al., 2001), was due to an effect of the antibodies on the post‐ganglionic sympathetic neurons. Equally, the hyponatraemia could be due to hypersecretion of antidiuretic hormone, and similar effects on other hypothalamic peptides could be responsible for the hyperphagia and weight gain in Cases 9 and 10. Although both cases reported by Buckley et al. (2001) had increased secretions, and it was suggested that this might be characteristic feature of VGKC‐Ab‐associated conditions, as nine out of the 10 patients described here did not have these symptoms it is unlikely to be a useful clinical hallmark in this condition.
The detection of 10 patients with this clinical syndrome, presenting mainly at two centres during a 15‐month period, suggests that it may be relatively common. Indeed, during the same period, only one case of paraneoplastic limbic encephalitis was seen (a case of testicular cancer with Ma2 antibodies). It is possible that this non‐paraneoplastic form of immune‐mediated encephalitis is presently misdiagnosed and thus inappropriately managed. Patients presenting with an acute or subacute encephalopathic illness are a common diagnostic problem facing general physicians and neurologists alike. Herpes zoster infection and Korsakoff’s syndrome remain two of the most important diagnoses to consider because specific treatments exist and there may be a poor outcome if treatment is delayed. Indeed many patients are started on aciclovir and thiamine replacement on admission, awaiting further investigation. Even if viral PCR is negative, a viral‐related condition is usually presumed, particularly if paraneoplastic antibodies are absent. Another possibility is Hashimoto’s encephalopathy, and since there are now doubts about the relevance of thyroid antibodies in Hashimoto’s encephalopathy (Chong et al., 2003), it would be worth testing patients in whom this diagnosis is entertained for VGKC‐Abs. Of practical use is the fact that, although the VGKC‐Ab assay is usually performed by immunoprecipitation, high titres of these antibodies may be identifiable during routine paraneoplastic testing on the basis of their specific pattern of binding to cerebellar sections (Fig. 4).
In some of the cases we describe, there was a marked decline in VGKC‐Ab levels, and the antibody levels appeared to remain low even when treatment was tailed off (Fig. 1B). This decline in antibody levels appears faster than that seen during conventional treatment of other autoimmune conditions, such as myasthenia gravis. In addition, two of the fastest responding patients described here (Cases 2 and 4) had a flu‐like illness preceding their condition, and two others had noted preceding infections. Moreover, a previous non‐paraneoplastic case (Buckley et al., 2001) improved spontaneously as her VGKC‐Abs spontaneously fell over a period of 2 years. Together, these observations suggest that some of these cases may have a monophasic illness that could be post‐infectious, with a natural time course perhaps somewhat similar to that of Guillain–Barré syndrome (Yuki et al., 2000; Press et al., 2001). In some cases, however, VGKC‐Abs tended to remain high despite treatment, suggesting a different natural history to the disease. However, it is too soon to say whether these differences are real or reflect the results of variable treatment regimens.
The issue of how to treat these patients effectively cannot be adequately addressed in this retrospective study. Whilst it is possible that VGKC‐Ab levels would decline in all patients with time, it would seem desirable to reduce their titre as fast as possible to stop seizure activity and with the aim of preventing permanent cerebral atrophy and disability. Although some patients showed a dramatic response to plasma exchange or IvIg, most did not, but most improved after a few weeks of prednisolone. This may prove to be why Case 8, who only started corticosteroids after a long period during which symptoms were unchanged, is now beginning to show some improvement (data not shown). It may also be that sustained use of oral prednisolone will prove to be more beneficial than, for instance, a short course of intravenous dexamethasone as used in Case 10. Even in a well‐defined autoantibody‐mediated disease such as myasthenia gravis, the response to IvIg or plasma exchange is less than complete (Gajdos et al., 1997), and 4–6 months of corticosteroids may be required to induce a remission, often associated with smaller falls in acetyl choline receptor (AChR) antibody levels (Palace et al., 1998) than the decline in VGKC‐Abs shown here. Whilst it is possible that such treatments work more effectively in a peripheral disease than in a central nervous system disorder, immunosuppression could act not only by reducing serum antibody levels, and thus the amount of antibody that can enter the CNS, but also by reducing inflammation and permeability of the blood–brain barrier (e.g. Gaillard et al., 2001). On the limited evidence available, we suggest that this condition is treated initially with IvIg or plasma exchange to try to obtain a quick clinical response, followed by high‐dose prednisolone for at least 6 months. If the antibody remains high, alternative immunosuppression may be useful. When the patient begins to improve, the dose of immunosuppressive drugs should be maintained until the improvement has stabilized. Further studies using a variety of outcome measures (including imaging, neuropsychology and antibody titres) will help to determine optimum therapy. In addition, animal models of the condition may throw light on the mechanisms of VGKC‐Ab‐associated encephalopathy and the effects of different treatments.
The diagnosis of VGKC‐Ab‐associated limbic encephalitis should be suspected in patients of either sex presenting with subacute onset of disorientation, confusion and memory loss particularly when associated with medial temporal lobe signal change on MRI. Clinically, these cases do not differ substantially from other forms of amnesic encephalopathies, such as the paraneoplastic form of limbic encephalitis associated with small‐cell lung cancer and Hu antibodies (Gultekin et al., 2000), except in the relative absence of cerebellar and brainstem involvement. However, VGKC‐Ab‐associated encephalopathy may have a wider phenotype, and seizures or more florid psychiatric symptoms such as hallucinations, may be the presenting features in some cases, including some with VGKC‐Abs in the 100–400 pM range (J. Palace, B. Lang et al., in preparation). The prompt recognition and treatment of these conditions may prevent the mortality associated with intractable seizures and electrolyte disturbances, and the morbidity associated with cerebral atrophy.
Acknowledgements
We wish to thank Dr John Stevens for radiological advice, and Drs Geoff Schott, Nigel Hyman and Dennis Briley for allowing us to study their cases. This work has been supported partly by the Alzheimer’s Society (UK) (J.M.S.), the Wellcome Trust (B.L.) and the Muscular Dystrophy Campaign (L.C.).
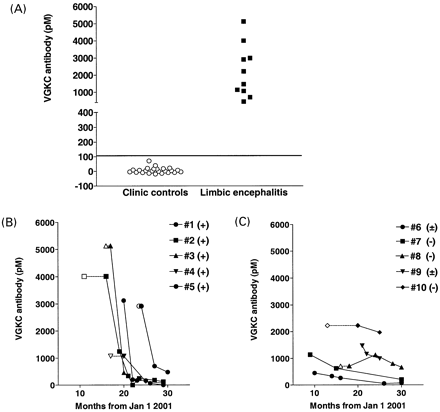
Fig. 1 (A) VGKC‐Abs in limbic encephalitis patients and in controls. Elevated levels of VGKC‐Abs were not found in 24 sera from unselected patients attending a DGH out‐patient clinic but were present in 10 patients with symptoms of limbic encephalitis. (B) VGKC‐Abs over time in patients showing a marked fall in antibody; all of these patients were given steroids (+) relatively early during the course of the disease. (C) VGKC‐Abs over time in patients showing a slower fall in antibody; in four of these patients, corticosteroid treatment was delayed (±) or not given (–). Open symbols and fine lines indicate the period from first symptoms to first antibody sampling.
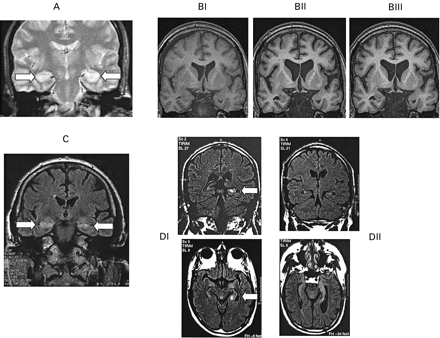
Fig. 2 MRI. (A) Case 1: T2‐weighted MRI showing bilateral hippocampal signal change (arrows) at disease onset. (B) Case 2: T1‐weighted coronal MRIs are shown from three time‐points: (BI) April 2002; (BII) October 2002; and (BIII) April 2003. Between scans BI and BII marked global decline in cerebral volume definitely, but not exclusively, involving the medial temporal lobes. Scan BIII shows little, if any, progression relative to BII, suggesting that ongoing excess volume loss had declined or halted, coincident with the improvement in clinical state and subsequent to treatment with plasma exchange. Concurrent FLAIR (fluid‐attenuated inversion recovery) imaging (not shown) revealed initial medial temporal lobe signal change, which had reduced by scan BII, and was no longer present by scan BIII. (C) Case 8: FLAIR coronal MRI at onset is shown, revealing signal change affecting anterior temporal lobe structures including each hippocampus (arrows). (D) Case 10: coronal and axial FLAIR images showing increased signal of the left hippocampus (arrow) 6 months after the onset of symptoms (DI), and increased signal and volume loss in both hippocampi, more pronounced on the left (arrows), 6 months later (DII).
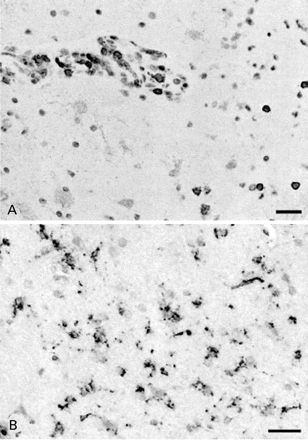
Fig. 3 Histopathology of the stereotactic biopsy of the left amygdala of Case 10. (A) Immunohistochemical staining for CD45 shows perivascular and parenchymal positive round cells (lymphocytes). (B) Staining for CD68 indicates diffuse microglial activation. Bars: 30 µm.
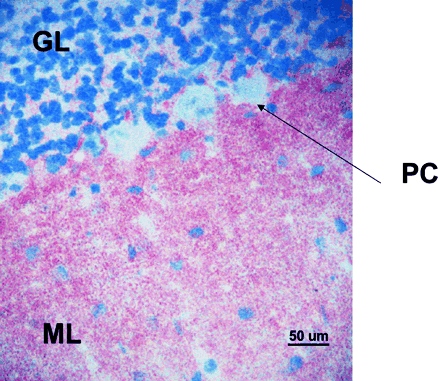
Fig. 4 Detection of VGKC‐Abs by routine indirect immunohistochemistry. All patients’ sera were negative for paraneoplastic antibodies but the five with the highest VGKC‐Ab titres (>2000 pM) produced a characteristic pink staining pattern on rat cerebellar neurons, binding strongly to the molecular layer and sparing the Purkinje cells. Two of the cases (1 and 3) were diagnosed during routine paraneoplastic screening by this characteristic staining pattern. PC = Purkinje cells; ML = molecular layer; GL = granular layer. Original magnification ×400.
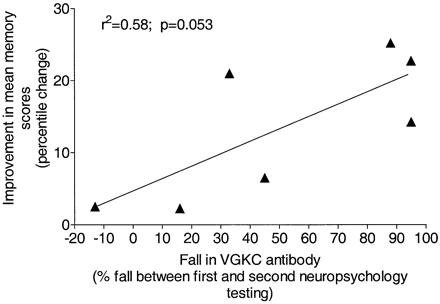
Fig. 5 Relationship between fall in VGKC‐Ab and clinical improvement. The difference between the mean percentile scores (mean of between two and six results for each patient; for details and comments see Table 3) for the memory tests before and after treatment are plotted against the percentage fall in VGKC‐Ab over the same time period for each patient.
Clinical features
| Patient | Time since firstsymptoms when studied by authors | Age at onset | Sex | Clinical presentation | Seizures | Other features | Neuromyotonia | Prodromal illness reported | |||||||||
| 1 | 1 week | 57 | M | M, C, S | G | H, Co, neuromyotonia, sweating | Yes | None | |||||||||
| 2 | 5 months | 57 | M | M, C | G, P | B, D (to phenytoin) ITU (Chest infection) | No | Flu‐like | |||||||||
| 3 | 3 weeks | 79 | F | M, C | G, P | H | No | None | 4 | 3 months | 71 | M | S, C | G | No | Flu like, weight loss | |
| 5 | 4 weeks | 76 | M | C, M, S | G | Gait disturbance, B, H ITU (septicaemia) | No | None | |||||||||
| 6 | 8 months | 59 | M | M, C, B | None | Gait dist, Co | No | Depression | |||||||||
| 7 | 2 weeks | 54 | M | C, M | P | B | No | Eye infection | |||||||||
| 8 | 6 weeks | 64 | M | M, C | G, P | D (to phenytoin) ITU, I | No | Diarrhoea | |||||||||
| 9 | 1 year | 44 | M | M | P | B, Co | No | None | |||||||||
| 10 | 7 months | 56 | M | C, M | P | Anxiety, delusions | No | None |
| Patient | Time since firstsymptoms when studied by authors | Age at onset | Sex | Clinical presentation | Seizures | Other features | Neuromyotonia | Prodromal illness reported | |||||||||
| 1 | 1 week | 57 | M | M, C, S | G | H, Co, neuromyotonia, sweating | Yes | None | |||||||||
| 2 | 5 months | 57 | M | M, C | G, P | B, D (to phenytoin) ITU (Chest infection) | No | Flu‐like | |||||||||
| 3 | 3 weeks | 79 | F | M, C | G, P | H | No | None | 4 | 3 months | 71 | M | S, C | G | No | Flu like, weight loss | |
| 5 | 4 weeks | 76 | M | C, M, S | G | Gait disturbance, B, H ITU (septicaemia) | No | None | |||||||||
| 6 | 8 months | 59 | M | M, C, B | None | Gait dist, Co | No | Depression | |||||||||
| 7 | 2 weeks | 54 | M | C, M | P | B | No | Eye infection | |||||||||
| 8 | 6 weeks | 64 | M | M, C | G, P | D (to phenytoin) ITU, I | No | Diarrhoea | |||||||||
| 9 | 1 year | 44 | M | M | P | B, Co | No | None | |||||||||
| 10 | 7 months | 56 | M | C, M | P | Anxiety, delusions | No | None |
M = memory impairment; C = confusion and disorientation; S = seizures; B = behaviour change; G = grand mal; P = partial; H = hallucinations; B, Behaviour change; Co = confabulation; D = drug reaction; ITU = ITU admission.
Clinical features
| Patient | Time since firstsymptoms when studied by authors | Age at onset | Sex | Clinical presentation | Seizures | Other features | Neuromyotonia | Prodromal illness reported | |||||||||
| 1 | 1 week | 57 | M | M, C, S | G | H, Co, neuromyotonia, sweating | Yes | None | |||||||||
| 2 | 5 months | 57 | M | M, C | G, P | B, D (to phenytoin) ITU (Chest infection) | No | Flu‐like | |||||||||
| 3 | 3 weeks | 79 | F | M, C | G, P | H | No | None | 4 | 3 months | 71 | M | S, C | G | No | Flu like, weight loss | |
| 5 | 4 weeks | 76 | M | C, M, S | G | Gait disturbance, B, H ITU (septicaemia) | No | None | |||||||||
| 6 | 8 months | 59 | M | M, C, B | None | Gait dist, Co | No | Depression | |||||||||
| 7 | 2 weeks | 54 | M | C, M | P | B | No | Eye infection | |||||||||
| 8 | 6 weeks | 64 | M | M, C | G, P | D (to phenytoin) ITU, I | No | Diarrhoea | |||||||||
| 9 | 1 year | 44 | M | M | P | B, Co | No | None | |||||||||
| 10 | 7 months | 56 | M | C, M | P | Anxiety, delusions | No | None |
| Patient | Time since firstsymptoms when studied by authors | Age at onset | Sex | Clinical presentation | Seizures | Other features | Neuromyotonia | Prodromal illness reported | |||||||||
| 1 | 1 week | 57 | M | M, C, S | G | H, Co, neuromyotonia, sweating | Yes | None | |||||||||
| 2 | 5 months | 57 | M | M, C | G, P | B, D (to phenytoin) ITU (Chest infection) | No | Flu‐like | |||||||||
| 3 | 3 weeks | 79 | F | M, C | G, P | H | No | None | 4 | 3 months | 71 | M | S, C | G | No | Flu like, weight loss | |
| 5 | 4 weeks | 76 | M | C, M, S | G | Gait disturbance, B, H ITU (septicaemia) | No | None | |||||||||
| 6 | 8 months | 59 | M | M, C, B | None | Gait dist, Co | No | Depression | |||||||||
| 7 | 2 weeks | 54 | M | C, M | P | B | No | Eye infection | |||||||||
| 8 | 6 weeks | 64 | M | M, C | G, P | D (to phenytoin) ITU, I | No | Diarrhoea | |||||||||
| 9 | 1 year | 44 | M | M | P | B, Co | No | None | |||||||||
| 10 | 7 months | 56 | M | C, M | P | Anxiety, delusions | No | None |
M = memory impairment; C = confusion and disorientation; S = seizures; B = behaviour change; G = grand mal; P = partial; H = hallucinations; B, Behaviour change; Co = confabulation; D = drug reaction; ITU = ITU admission.
Investigations
| Patient | Low Na+ | CSF WCC | CSF protein (g/l) | Oligoclonal bands* | Brain MRI | EEG | EMG | Initial VGKC‐Ab (pM) [serum, CSF (CSF/serum)] |
| 1 | Yes | 11 | 0.75 | Matched serum/CSF | BM | N | NMT | 3120, 350 (11 %) |
| 2 | Yes | <4 | 0.5 | Matched serum/CSF | BM | D, F | Normal | 4005, 133 (3 %) |
| 3 | Yes | <4 | <0.4 | Insufficient sample | UM (L) | D, F | Normal | 5128 |
| 4 | Yes | <4 | <0.4 | Not available | UM (L) | F, D | Normal | 1232 |
| 5 | Yes | <4 | <0.4 | Matched serum/CSF | N | D, F | Normal | 4712, 71 (1.5 %) |
| 6 | Yes | 8 | <0.4 | Unmatched | N | N | Not done | 450, <10 (<2 %) |
| 7 | Yes | 6 | 0.8 | Negative | BM | F | Normal | 1139, 33 (3 %) |
| 8 | Yes | <4 | 0.42 | Matched serum/CSF | BM | D | Normal | 925 |
| 9 | No | 6 | 0.44 | Negative | BM | D, F | Normal | 1471 |
| 1 | No | 6 | 0.51 | Negative | UM (L) | F slowing | Not done | 2224 |
| Patient | Low Na+ | CSF WCC | CSF protein (g/l) | Oligoclonal bands* | Brain MRI | EEG | EMG | Initial VGKC‐Ab (pM) [serum, CSF (CSF/serum)] |
| 1 | Yes | 11 | 0.75 | Matched serum/CSF | BM | N | NMT | 3120, 350 (11 %) |
| 2 | Yes | <4 | 0.5 | Matched serum/CSF | BM | D, F | Normal | 4005, 133 (3 %) |
| 3 | Yes | <4 | <0.4 | Insufficient sample | UM (L) | D, F | Normal | 5128 |
| 4 | Yes | <4 | <0.4 | Not available | UM (L) | F, D | Normal | 1232 |
| 5 | Yes | <4 | <0.4 | Matched serum/CSF | N | D, F | Normal | 4712, 71 (1.5 %) |
| 6 | Yes | 8 | <0.4 | Unmatched | N | N | Not done | 450, <10 (<2 %) |
| 7 | Yes | 6 | 0.8 | Negative | BM | F | Normal | 1139, 33 (3 %) |
| 8 | Yes | <4 | 0.42 | Matched serum/CSF | BM | D | Normal | 925 |
| 9 | No | 6 | 0.44 | Negative | BM | D, F | Normal | 1471 |
| 1 | No | 6 | 0.51 | Negative | UM (L) | F slowing | Not done | 2224 |
Paraneoplastic antibodies were negative in all patients, but Case 8 had antinuclear antibodies. *Unmatched bands, indicate an expanded population of antibodies in the CSF that reflect intrathecal synthesis; matched bands in serum and CSF indicate an expanded population of antibodies in both serum and CSF. WCC = white cell count; BM = bilateral medial temporal lobe signal change; UM = unilateral medial temporal lobe signal change; N = normal; UB = unilateral basal ganglia signal change; D = diffuse slowing; F = focal activity.
Investigations
| Patient | Low Na+ | CSF WCC | CSF protein (g/l) | Oligoclonal bands* | Brain MRI | EEG | EMG | Initial VGKC‐Ab (pM) [serum, CSF (CSF/serum)] |
| 1 | Yes | 11 | 0.75 | Matched serum/CSF | BM | N | NMT | 3120, 350 (11 %) |
| 2 | Yes | <4 | 0.5 | Matched serum/CSF | BM | D, F | Normal | 4005, 133 (3 %) |
| 3 | Yes | <4 | <0.4 | Insufficient sample | UM (L) | D, F | Normal | 5128 |
| 4 | Yes | <4 | <0.4 | Not available | UM (L) | F, D | Normal | 1232 |
| 5 | Yes | <4 | <0.4 | Matched serum/CSF | N | D, F | Normal | 4712, 71 (1.5 %) |
| 6 | Yes | 8 | <0.4 | Unmatched | N | N | Not done | 450, <10 (<2 %) |
| 7 | Yes | 6 | 0.8 | Negative | BM | F | Normal | 1139, 33 (3 %) |
| 8 | Yes | <4 | 0.42 | Matched serum/CSF | BM | D | Normal | 925 |
| 9 | No | 6 | 0.44 | Negative | BM | D, F | Normal | 1471 |
| 1 | No | 6 | 0.51 | Negative | UM (L) | F slowing | Not done | 2224 |
| Patient | Low Na+ | CSF WCC | CSF protein (g/l) | Oligoclonal bands* | Brain MRI | EEG | EMG | Initial VGKC‐Ab (pM) [serum, CSF (CSF/serum)] |
| 1 | Yes | 11 | 0.75 | Matched serum/CSF | BM | N | NMT | 3120, 350 (11 %) |
| 2 | Yes | <4 | 0.5 | Matched serum/CSF | BM | D, F | Normal | 4005, 133 (3 %) |
| 3 | Yes | <4 | <0.4 | Insufficient sample | UM (L) | D, F | Normal | 5128 |
| 4 | Yes | <4 | <0.4 | Not available | UM (L) | F, D | Normal | 1232 |
| 5 | Yes | <4 | <0.4 | Matched serum/CSF | N | D, F | Normal | 4712, 71 (1.5 %) |
| 6 | Yes | 8 | <0.4 | Unmatched | N | N | Not done | 450, <10 (<2 %) |
| 7 | Yes | 6 | 0.8 | Negative | BM | F | Normal | 1139, 33 (3 %) |
| 8 | Yes | <4 | 0.42 | Matched serum/CSF | BM | D | Normal | 925 |
| 9 | No | 6 | 0.44 | Negative | BM | D, F | Normal | 1471 |
| 1 | No | 6 | 0.51 | Negative | UM (L) | F slowing | Not done | 2224 |
Paraneoplastic antibodies were negative in all patients, but Case 8 had antinuclear antibodies. *Unmatched bands, indicate an expanded population of antibodies in the CSF that reflect intrathecal synthesis; matched bands in serum and CSF indicate an expanded population of antibodies in both serum and CSF. WCC = white cell count; BM = bilateral medial temporal lobe signal change; UM = unilateral medial temporal lobe signal change; N = normal; UB = unilateral basal ganglia signal change; D = diffuse slowing; F = focal activity.
Neuropsychological scores before treatment
| Case | NART | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 110 | 115 | 110 | 5th | <5th | 3rd | <1st | >16th | <1st | <1st | |||
| 2 | 72 | 61 | 65 | <5th | <5th | <5th | |||||||
| 4 | 101 | 91 | 88 | <1st | <1st | <1st | <1st | 11th–16th | 16th | 2nd | |||
| 6 | 89 | 19th | 12th | 38th | 4th | >16th | 86th | 76th | |||||
| 7 | 113 | 115 | 112 | 5th | 1st | 1st | 1st | 2nd–5th | <1st | 1st | |||
| 8 | 81 | 84 | 86 | <5th | <5th | 37th | |||||||
| 9 | 107 | 98 | 102 | 28th | 1st | 25th | 5th | >16th | 34th | 63rd |
| Case | NART | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 110 | 115 | 110 | 5th | <5th | 3rd | <1st | >16th | <1st | <1st | |||
| 2 | 72 | 61 | 65 | <5th | <5th | <5th | |||||||
| 4 | 101 | 91 | 88 | <1st | <1st | <1st | <1st | 11th–16th | 16th | 2nd | |||
| 6 | 89 | 19th | 12th | 38th | 4th | >16th | 86th | 76th | |||||
| 7 | 113 | 115 | 112 | 5th | 1st | 1st | 1st | 2nd–5th | <1st | 1st | |||
| 8 | 81 | 84 | 86 | <5th | <5th | 37th | |||||||
| 9 | 107 | 98 | 102 | 28th | 1st | 25th | 5th | >16th | 34th | 63rd |
Neuropsychological scores before treatment
| Case | NART | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 110 | 115 | 110 | 5th | <5th | 3rd | <1st | >16th | <1st | <1st | |||
| 2 | 72 | 61 | 65 | <5th | <5th | <5th | |||||||
| 4 | 101 | 91 | 88 | <1st | <1st | <1st | <1st | 11th–16th | 16th | 2nd | |||
| 6 | 89 | 19th | 12th | 38th | 4th | >16th | 86th | 76th | |||||
| 7 | 113 | 115 | 112 | 5th | 1st | 1st | 1st | 2nd–5th | <1st | 1st | |||
| 8 | 81 | 84 | 86 | <5th | <5th | 37th | |||||||
| 9 | 107 | 98 | 102 | 28th | 1st | 25th | 5th | >16th | 34th | 63rd |
| Case | NART | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 110 | 115 | 110 | 5th | <5th | 3rd | <1st | >16th | <1st | <1st | |||
| 2 | 72 | 61 | 65 | <5th | <5th | <5th | |||||||
| 4 | 101 | 91 | 88 | <1st | <1st | <1st | <1st | 11th–16th | 16th | 2nd | |||
| 6 | 89 | 19th | 12th | 38th | 4th | >16th | 86th | 76th | |||||
| 7 | 113 | 115 | 112 | 5th | 1st | 1st | 1st | 2nd–5th | <1st | 1st | |||
| 8 | 81 | 84 | 86 | <5th | <5th | 37th | |||||||
| 9 | 107 | 98 | 102 | 28th | 1st | 25th | 5th | >16th | 34th | 63rd |
Neuropsychological scores after treatment and relation with VGKC‐Ab titres
| Case | Time (months) and VGKC‐Ab [change (%) between tests] | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 3, –95 | 115 | 107 | 48th | 41st | 15th | <1st | 85th | 94th | ||||
| 2 | 12, –95 | 90 | 114 | 36th | 11th | 25th | 5st | >16th | 24th | 63rd | |||
| 4 | 6, –88 | 98 | 110 | 75th | 20th | 9th | 1st | >16th | 31st | 14th | |||
| 6*,† | 4, –16 | 37th | 37th | 3rd | 5th | >16th | 90th | 86th | |||||
| 7† | 0.5, –45 | 16th | 1st | 1st | <1st | ||||||||
| 8 | 10, +13 | 83 | 86 | 10th | <5th | 37th | |||||||
| 9 | 5, –33 | 112 | 111 | 54th | 36th | 37th | 16th | >16th | 16th | 75rd |
| Case | Time (months) and VGKC‐Ab [change (%) between tests] | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 3, –95 | 115 | 107 | 48th | 41st | 15th | <1st | 85th | 94th | ||||
| 2 | 12, –95 | 90 | 114 | 36th | 11th | 25th | 5st | >16th | 24th | 63rd | |||
| 4 | 6, –88 | 98 | 110 | 75th | 20th | 9th | 1st | >16th | 31st | 14th | |||
| 6*,† | 4, –16 | 37th | 37th | 3rd | 5th | >16th | 90th | 86th | |||||
| 7† | 0.5, –45 | 16th | 1st | 1st | <1st | ||||||||
| 8 | 10, +13 | 83 | 86 | 10th | <5th | 37th | |||||||
| 9 | 5, –33 | 112 | 111 | 54th | 36th | 37th | 16th | >16th | 16th | 75rd |
NART = National Adult Reading Test or Schonell Graded Reading Test predicted VIQ; VIQ = verbal IQ; PIQ = performance IQ; AMIPB SR = Adult Memory and Information Processing Battery: Story Recall; AMIPB LL = Adult Memory and Information Processing Battery: List Learning; RMT = Warrington Recognition Memory Test; Rey CFT = Rey Complex Figure Test; GNT = Graded Naming Test; IR = immediate recall; DR = delayed recall; W = words; F = faces. Note: all scores are percentiles except for NART, VIQ and PIQ. VGKC‐Ab titres are given as a percentage change between the pre‐ and post‐treatment values. *For Case 6, some improvement had already been noted before the first neuropsychology testing. †For Cases 6 and 7 the VGKC‐Ab testing was not well matched to the times of the neuropsychological tests.
Neuropsychological scores after treatment and relation with VGKC‐Ab titres
| Case | Time (months) and VGKC‐Ab [change (%) between tests] | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 3, –95 | 115 | 107 | 48th | 41st | 15th | <1st | 85th | 94th | ||||
| 2 | 12, –95 | 90 | 114 | 36th | 11th | 25th | 5st | >16th | 24th | 63rd | |||
| 4 | 6, –88 | 98 | 110 | 75th | 20th | 9th | 1st | >16th | 31st | 14th | |||
| 6*,† | 4, –16 | 37th | 37th | 3rd | 5th | >16th | 90th | 86th | |||||
| 7† | 0.5, –45 | 16th | 1st | 1st | <1st | ||||||||
| 8 | 10, +13 | 83 | 86 | 10th | <5th | 37th | |||||||
| 9 | 5, –33 | 112 | 111 | 54th | 36th | 37th | 16th | >16th | 16th | 75rd |
| Case | Time (months) and VGKC‐Ab [change (%) between tests] | VIQ | PIQ | AMIPB SR | AMIPB LL | RMT | Rey CFT | GNT | |||||
| IR | DR | A1‐5 | A6 | W | F | Copy | IR | DR | |||||
| 1 | 3, –95 | 115 | 107 | 48th | 41st | 15th | <1st | 85th | 94th | ||||
| 2 | 12, –95 | 90 | 114 | 36th | 11th | 25th | 5st | >16th | 24th | 63rd | |||
| 4 | 6, –88 | 98 | 110 | 75th | 20th | 9th | 1st | >16th | 31st | 14th | |||
| 6*,† | 4, –16 | 37th | 37th | 3rd | 5th | >16th | 90th | 86th | |||||
| 7† | 0.5, –45 | 16th | 1st | 1st | <1st | ||||||||
| 8 | 10, +13 | 83 | 86 | 10th | <5th | 37th | |||||||
| 9 | 5, –33 | 112 | 111 | 54th | 36th | 37th | 16th | >16th | 16th | 75rd |
NART = National Adult Reading Test or Schonell Graded Reading Test predicted VIQ; VIQ = verbal IQ; PIQ = performance IQ; AMIPB SR = Adult Memory and Information Processing Battery: Story Recall; AMIPB LL = Adult Memory and Information Processing Battery: List Learning; RMT = Warrington Recognition Memory Test; Rey CFT = Rey Complex Figure Test; GNT = Graded Naming Test; IR = immediate recall; DR = delayed recall; W = words; F = faces. Note: all scores are percentiles except for NART, VIQ and PIQ. VGKC‐Ab titres are given as a percentage change between the pre‐ and post‐treatment values. *For Case 6, some improvement had already been noted before the first neuropsychology testing. †For Cases 6 and 7 the VGKC‐Ab testing was not well matched to the times of the neuropsychological tests.
Treatments, serological and clinical responses
| Patient | Plasma exchange | IvIg | Steroids | VGKC‐Ab (pM) at June 2003 (% initial value) | Clinical response | Persistent defects | Follow up MRI |
| 1 | + | + | + | 63 (2) | Definite | M | Resolution of high signal and bilateral atrophy |
| 2 | + | + | + | 124 (3) | Definite | M | Atrophy |
| 3 | – | + (3×) | + | 459 (9) | Definite | M relapses | Reduced signal change in hipppocampus |
| 4 | – | + | + | 131 (11) | Definite | M | Normal |
| 5 | + | + (3×) | + (+ Aza) | 480 (16) | Definite | M | Normal |
| 6 | + | – | + delayed | 77 (17) | Definite | M, P | Bilateral hippocampal atrophy |
| 7 | + | + (2×) | – | 205 (18) | None | M, P | Normal |
| 8 | + | + | – | 665 (58) | Slight | M, P, S | Persistent atrophy |
| 9 | + | – | + delayed | 941 (67) | Slight | M, P | Normal |
| 10 | – | – | Intravenous dexamethasone one course only | 1968 (88) | Slight | M | Bilateral hippocampal atrophy and signal change |
| Patient | Plasma exchange | IvIg | Steroids | VGKC‐Ab (pM) at June 2003 (% initial value) | Clinical response | Persistent defects | Follow up MRI |
| 1 | + | + | + | 63 (2) | Definite | M | Resolution of high signal and bilateral atrophy |
| 2 | + | + | + | 124 (3) | Definite | M | Atrophy |
| 3 | – | + (3×) | + | 459 (9) | Definite | M relapses | Reduced signal change in hipppocampus |
| 4 | – | + | + | 131 (11) | Definite | M | Normal |
| 5 | + | + (3×) | + (+ Aza) | 480 (16) | Definite | M | Normal |
| 6 | + | – | + delayed | 77 (17) | Definite | M, P | Bilateral hippocampal atrophy |
| 7 | + | + (2×) | – | 205 (18) | None | M, P | Normal |
| 8 | + | + | – | 665 (58) | Slight | M, P, S | Persistent atrophy |
| 9 | + | – | + delayed | 941 (67) | Slight | M, P | Normal |
| 10 | – | – | Intravenous dexamethasone one course only | 1968 (88) | Slight | M | Bilateral hippocampal atrophy and signal change |
M = memory deficit; P = personality change; S = seizures.
Treatments, serological and clinical responses
| Patient | Plasma exchange | IvIg | Steroids | VGKC‐Ab (pM) at June 2003 (% initial value) | Clinical response | Persistent defects | Follow up MRI |
| 1 | + | + | + | 63 (2) | Definite | M | Resolution of high signal and bilateral atrophy |
| 2 | + | + | + | 124 (3) | Definite | M | Atrophy |
| 3 | – | + (3×) | + | 459 (9) | Definite | M relapses | Reduced signal change in hipppocampus |
| 4 | – | + | + | 131 (11) | Definite | M | Normal |
| 5 | + | + (3×) | + (+ Aza) | 480 (16) | Definite | M | Normal |
| 6 | + | – | + delayed | 77 (17) | Definite | M, P | Bilateral hippocampal atrophy |
| 7 | + | + (2×) | – | 205 (18) | None | M, P | Normal |
| 8 | + | + | – | 665 (58) | Slight | M, P, S | Persistent atrophy |
| 9 | + | – | + delayed | 941 (67) | Slight | M, P | Normal |
| 10 | – | – | Intravenous dexamethasone one course only | 1968 (88) | Slight | M | Bilateral hippocampal atrophy and signal change |
| Patient | Plasma exchange | IvIg | Steroids | VGKC‐Ab (pM) at June 2003 (% initial value) | Clinical response | Persistent defects | Follow up MRI |
| 1 | + | + | + | 63 (2) | Definite | M | Resolution of high signal and bilateral atrophy |
| 2 | + | + | + | 124 (3) | Definite | M | Atrophy |
| 3 | – | + (3×) | + | 459 (9) | Definite | M relapses | Reduced signal change in hipppocampus |
| 4 | – | + | + | 131 (11) | Definite | M | Normal |
| 5 | + | + (3×) | + (+ Aza) | 480 (16) | Definite | M | Normal |
| 6 | + | – | + delayed | 77 (17) | Definite | M, P | Bilateral hippocampal atrophy |
| 7 | + | + (2×) | – | 205 (18) | None | M, P | Normal |
| 8 | + | + | – | 665 (58) | Slight | M, P, S | Persistent atrophy |
| 9 | + | – | + delayed | 941 (67) | Slight | M, P | Normal |
| 10 | – | – | Intravenous dexamethasone one course only | 1968 (88) | Slight | M | Bilateral hippocampal atrophy and signal change |
M = memory deficit; P = personality change; S = seizures.
References
Alamowitch S, Graus F, Uchuya M, Rene R, Bescansa E, Delattre JY. Limbic encephalitis and small cell lung cancer. Clinical and immunological features.
Amyes E, Curnow J, Stark Z, Corlett L, Sutton I, Vincent A. Restricted IgG1 subclass of anti‐Yo antibodies in paraneoplastic cerebellar degeneration.
Antoine JC, Honnorat J, Anterion CT, Aguera M, Absi L, Fournel P, et al. Limbic encephalitis and immunological perturbations in two patients with thymoma.
Bak TH, Antoun N, Balan KK, Hodges JR. Memory lost, memory regained: neuropsychological findings and neuroimaging in two cases of paraneoplastic limbic encephalitis with radically different outcomes.
Barber PA, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage‐gated K+ channel antibodies.
Bien CG, Schulze‐Bonhage A, Deckert M, Urbach H, Helmstaedter C, Grunwald T, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy.
Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas.
Buckley C, Oger J, Clover L, Tuzun E, Carpenter K, Jackson M, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis.
Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth?
Corsellis JA, Goldberg GJ, Norton AR. ‘Limbic encephalitis’ and its association with carcinoma.
Coughlan AK, Hollows SE. The adult memory and information processing battery. Leeds: A. K. Coughlan, Psychology Department, St James University Hospital;
Dalmau J, Graus F, Rosenblum MK, Posner JB. Anti‐Hu‐associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients.
Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology.
Gaillard PJ, van Der Meide PH, de Boer AG, Breimer DD. Glucocorticoid and type 1 interferon interactions at the blood–brain barrier: relevance for drug therapies for multiple sclerosis.
Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high‐dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group.
Gratacos E, Ghelardini C, Gherardini LM, Galeotti N, Murphy KJ, Bartolini A, et al. Kv1.1 channel antisense attenuates learning and modulation of dentate polysialylated NCAM.
Graus F, Keime‐Guibert F, Rene R, Benyahia B, Ribalta T, Ascaso C, et al. Anti‐Hu‐associated paraneoplastic encephalomyelitis: analysis of 200 patients.
Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients.
Hart IK, Waters C, Vincent A, Newland C, Beeson D, Pongs O, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia.
Hart IK, Maddison P, Newsom‐Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability.
Lee EK, Maselli RA, Ellis WG, Agius MA. Morvan’s fibrillary chorea: a paraneoplastic manifestation of thymoma.
Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage‐gated potassium channels.
Lishman WA. Organic psychiatry. The psychological consequences of cerebral disorder. 3rd ed. Oxford: Blackwell Science;
Monaghan MM, Trimmer JS, Rhodes KJ. Experimental localization of Kv1 family voltage‐gated K+ channel alpha and beta subunits in rat hippocampal formation.
Mori M, Kuwabara S, Yoshiyama M, Kanesaka T, Ogata T, Hattori T. Successful immune treatment for non‐paraneoplastic limbic encephalitis.
Palace J, Newsom‐Davis J, Lecky B. A randomized double‐blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group.
Palmer BF. Hyponatremia in patients with central nervous system disease: SIADH versus CSW.
Pozo‐Rosich P, Clover L, Saiz A, Vincent A, Graus F. Voltage‐gated potassium channel antibodies in limbic encephalitis.
Press R, Mata S, Lolli F, Zhu J, Andersson T, Link H. Temporal profile of anti‐ganglioside antibodies and their relation to clinical parameters and treatment in Guillain–Barre syndrome.
Rhodes KJ, Monagham MM, Barrezueta NX, Nawoschik S, Bekele‐Arcuri Z, Matos MF, et al. Voltage‐gated K+ channel beta subunits: expression and distribution of Kvbeta1 and Kvbeta2 in adult rat brain.
Rosenfeld MR, Eichen JG, Wade DF, Posner JB, Dalmau J. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins.
Schott JM, Harkness K, Barnes J, della Rocchetta AI, Vincent A, Rossor MN. Amnesia, cerebral atrophy, and autoimmunity.
Shillito P, Molenaar PC, Vincent A, Leys K, Zheng W, van den Berg RJ, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves.
Vernino S, Lennon VA. Ion channel and striational antibodies define a continuum of autoimmune neuromuscular hyperexcitability.
Vincent A. Measuring and evaluating the significance of autoantibodies in neurological disorders.
Voltz R, Gultekin SH, Rosenfeld MR, Gerstner E, Eichen J, Posner JB, et al. A serologic marker of paraneoplastic limbic and brain‐stem encephalitis in patients with testicular cancer.
Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain.
Wechsler DA. Wechsler Adult Intelligence Scale: Revised Manual. New York: Psychological Corporation;
Yuki N, Ang CW, Koga M, Jacobs BC, van Doorn PA, Hirata K, et al. Clinical features and response to treatment in Guillain–Barre syndrome associated with antibodies to GM1B ganglioside.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}