




Abstract
We describe a pedigree of Anglo‐Celtic origin with a phenotypically unique form of dominantly inherited spinocerebellar ataxia (SCA) in 14 personally examined affected members. A remarkable observation is dentate nucleus calcification, producing a low signal on MRI sequences. Unusually for an SCA, dysarthria is typically the initial manifestation. Mild pyramidal signs and hypermetric saccades are noted in some. Its distinguishing clinical features, each present in a majority of affected persons, are palatal tremor, and a form of dysphonia resembling spasmodic dysphonia. Repeat expansion detection failed to identify either CAG/CTG or ATTCT/AGAAT repeat expansions segregating with the disease in this family. The testable SCA mutations have been excluded. On linkage analysis, the locus maps to chromosome 11, which rules out all the remaining mapped SCAs except for SCA5. While locus homogeneity with SCA5 is not formally excluded, we consider it rather unlikely on phenotypic grounds, and propose that this condition may represent an addition to the group of neurogenetic disorders subsumed under the rubric SCA. The International Nomenclature Committee has made a provisional assignment of ‘SCA20’, although firm designation will have to await a definite molecular distinction from SCA5.
Introduction
The autosomal dominant cerebellar ataxias (ADCAs) have been classified clinically according to the presence or absence of extracerebellar involvement, and genetically according to the demonstration of a specific mutation or a mapped locus. The genetic classification is based upon the demonstration of locus heterogeneity, and each newly identified ataxia is ascribed a spinocerebellar ataxia (SCA) number (Margolis, 2002; Chung et al., 2003). Many SCAs remain to be discovered (Subramony and Filla, 2001). Worldwide, about a third of the ADCA pedigrees remain unmapped (Zu et al., 1999), and a survey of pedigrees with dominantly inherited SCA in south‐eastern Australia showed that only about half were accounted for by the more common SCAs, namely SCAs 1, 2, 3, 6 and 7 (Storey et al., 2000). We have reviewed our neurogenetic clinic population to assess those families in which analysis might potentially be fruitful, and we report here the observations and findings from one such ADCA family. Notable additional clinical findings are dysphonia, resembling spasmodic dysphonia, which is superimposed on the cerebellar dysarthria, and palatal tremor. The striking correlate on neuroimaging is dentate nucleus calcification. The genetic rubric which has been applied following submission to the Human Gene Nomenclature Committee is SCA20, although formal proof of its distinction from SCA5, which maps to the same chromosomal region, is yet to be adduced.
Material and methods
Clinical studies on the family
The pedigree of the family is presented in Fig. 1. The propositus (III:11) was judged to have a spasmodic dysphonia superimposed upon his cerebellar dysarthria, and his report that other affected family members also had ‘squeaky voices’ led to further investigation of the pedigree. All known living affected family members were personally examined by the senior author (E.S.), neuroradiological examinations initiated, and previous studies reviewed. Formal voice analysis, using stroboscopic evaluation of vocal fold vibration and acoustic analysis of phonation of isolated vowels and connected speech samples, was undertaken in the propositus. Some affected persons had blood samples drawn for testing of calcium homeostasis. The study was approved by the Women’s and Children’s Hospital (Melbourne) Human Ethics Committee. All participants gave informed consent.
Genotyping and linkage analysis
Blood was collected from affected individuals and other appropriate and available family members for genotyping and direct gene testing, and genomic DNA prepared. The trinucleotide expansions of SCAs 1–3, 6–8, 17 and dentatorubral‐pallidoluysian atrophy (DRPLA) were tested in representative family members. Microsatellite markers reported in the literature were used to test linkage to the loci of SCAs 4–6 and 10–16, as follows: for the SCA4 locus at 16q22.1, markers D16S397 and D16S3050 (Flanigan et al., 1996; Nagaoka et al., 2000); the SCA5 locus at 11p12–q12, markers D11S903 and D11S913 (Ranum et al., 1994); the SCA6 locus, markers D19S221, D19S1150 and D19S226 (Yue et al., 1997); the SCA10 locus at 22q13, markers D22S274 and D22S928 (Zu et al., 1999); the SCA11 locus at 15q14–21.3, markers D15S994, D15S123 and D15S1039 (Worth et al., 1999); the SCA12 locus at 5q31–q33, markers D5S436 and D5S470; the SCA13 locus at 19q13.3–13.4, marker D19S902 (Herman‐Bert et al., 2000); the SCA14 locus at 19q13.3–qter, markers D19S206 and D19S605 (Yamashita et al., 2000); the SCA15 locus at 3p24.2–3pter, markers D3S3630, D3S3050 and D3S1560 (Knight et al., 2003); and for the SCA16 locus at 8q22.1–24.1, markers D8S514 and D8S284 (Miyoshi et al., 2001). Markers from the ABI PRISM Linkage Mapping Set, version 2 (PE Applied Biosystems), at a marker density of 10 centiMorgans (cM), were used for a genome‐wide scan. Fine‐mapping markers with respect to chromosome 11 were obtained from NCBI and GDB databases (National Center for Biotechnology Information; Genome Database).
Simulation studies using SIMLINK Version 4.1 (Ploughman and Boehnke, 1989) indicated that the family had the potential to demonstrate linkage, with a theoretical maximum logarithm of odds (LOD) score of 6.9 (average LOD score of 4.62), and would be capable of excluding linkage to 10.6 cM on either side of an unlinked marker. Transmission of SCA20 was assumed to be autosomal dominant, with estimates of age‐specific penetrance derived from a Kaplan–Meier plot of data from the affected persons (which gave an almost straight‐line graph from 0.1 at age 30 to 1.0 at 65 years). The disease allele was assigned a frequency of 0.0002 with 0% phenocopy rate. All markers with <10 alleles were given the equal allele frequency of 0.1, and markers with >10 alleles were given the frequency 1/(number of observed alleles). Equal rates of male and female recombination were assumed. Information on additional markers to confirm and refine the interval was accessed through the NCBI and GDB databases.
The data were prepared for input by genotyping error‐cleaning and pedigree checking with PREST (McPeek and Sun, 2000), PEDCHECK (O’Connell and Weeks, 1998) and UNKNOWN (Terwilliger and Ott, 1994). Genotyping errors were removed and one pedigree error corrected. Two‐point analysis was performed using the MLINK program of the LINKAGE package (version 5.1) (Lathrop et al., 1984). The Elston–Stewart algorithm in the form of the program VITESSE was applied for a limited multipoint analysis (5–10 marker blocks including the disease locus), using a single penetrance model with penetrances of Pr(disease|DD) = 0.0, Pr(disease|Dd) = 0.99, Pr(disease|dd) = 0.99.
Repeat expansion detection (RED) analysis
The RED technique was performed essentially as described in Schalling et al. (1993) to test for the presence of unstable CAG/CTG trinucleotide repeats, in samples from several family members. In addition to testing CAG/CTG repeats via RED, a modified version of the technique was used to investigate the ATTCT/AGAAT pentanucleotide repeat associated with SCA10 (Matsuura et al., 2000; Knight et al., 2003).
Results
Phenotype: clinical characterization of the SCA family
The pattern of transmission of the disease is consistent with autosomal dominant inheritance. A partial pedigree is shown in Fig. 1, restricted for the most part to showing those family members who could be assessed/sampled. In all, 14 affected members underwent systematic neurological history and examination (Table 1). The age at onset of first symptom, as self‐reported, ranged from 19 to 64 years, mean and median both 46.5 years. Three parent–child pairs (III:3/IV:2–3 and III:13/IV:5) were documented by self‐report with respect to age of onset, with an average of a 10 year earlier onset in the child. One member (IV:4) was judged to be possibly affected on clinical assessment at age 38 years, showing only postural/action tremor and impaired visual suppression of the vestibulo‐ocular reflex (VOR), but was shown subsequently on CT to have the characteristic dentate calcification (see below), confirming his status as affected. In generation IV (age range 30–50 years), among the 26 offspring of known affected persons, five were affected (Fig. 1), five we confirmed to be clinically unaffected, and the remaining 16 were anecdotally reported to be unaffected. None is reported to be affected in generation V, the oldest now entering their 30s. There was no instance of an affected offspring of an apparently unaffected parent.
The first symptom was dysarthria in nine of 14, gait ataxia in two, and both together in two. The dysarthria was reported to have been of sudden onset in two out of nine. The second symptom in those initially manifesting dysarthria was typically gait ataxia (seven out of nine), which followed from 3 months to 25 years later. In the other two out of nine, the second symptom was of upper limb ataxia (hammering nails, handwriting). In one out of 14, the only symptom was tremor. At the time of examination, illness duration had varied from 1 to 43 years. As judged by these cross‐sectional data, progression is probably slow, with one affected becoming wheelchair‐dependent after 40 years of symptoms, and one other requiring percutaneous gastrostomy feeding after 15 years of symptoms.
Upper limb ataxia typically was more prominent on ballistic tracking than ramp (slow finger/nose) movements (11 out of 14). Two affected members had a postural and action tremor affecting the head and arms, this being the only sign in one (IV:4). Index finger tapping rate was reduced (≥2 SD below normal for age, sex, education and hand) in at least one hand in seven out of 13 (IV:4 not tested). Plantar responses were flexor in 13 and equivocal in one, but five had minor pyramidal signs (hyper‐reflexic knee jerks without increased tone; crossed adductor responses). Sensation at the toes (vibration perception, two‐alternative forced choice method; pinprick; two‐point discrimination) was typically unremarkable, although three out of 14 had an increased two‐point discrimination threshold of >2 cm (i.e. greater than the diameter of their great toes).
Clinical examination of eye movements typically revealed hypermetric saccades into down gaze (10 out of 14), with eight also hypermetric on horizontal gaze. Smooth pursuit showed saccadic interruptions in three, while four had a mild or moderate excess of square wave jerks in the primary position. Nystagmus was typically absent, but two out of 14 showed slight, non‐sustained down and lateral nystagmus in down and lateral gaze (‘side pocket nystagmus’). The VOR gain was uniformly normal as judged by dynamic versus static visual acuity and by estimation of VOR gain by ophthalmoscopy during head oscillation (Zee, 1978). Visual suppression of the VOR was impaired in eight out of 14. Visual acuity and colour vision (Farnsworth‐Munsell 15D test) were normal in all, apart from III:8 who had a typical and presumably unrelated protanopia.
The clinical syndrome is thus of a relatively pure cerebellar ataxia, without prominent pyramidal features. In addition, two distinctive features were usually present. A dysphonia sounding superficially similar to adductor spasmodic dysphonia was present in nine out of 14, although in two of these it was mild, discernible on reading a prose paragraph but not in ordinary conversation. In those in whom it was more prominent, it was usually reported to have followed the development of dysarthria by several years. Palatal tremor of ∼2 Hz, unassociated with ear click, was seen in 10 out of 14; in two out of these 10, it was also evident in the lips.
Phenotype: imaging, clinical and laboratory investigations of the SCA family
CT showed pronounced dentate calcification in nine out of nine in whom scanning was done, including in three who had been symptomatic for ≤3 years, and in the one in whom the only clinical sign was tremor. In only two of these nine was there pallidal calcification, and in one of these it was very slight (IV:3). MRI showed mild to moderate pancerebellar atrophy, along with low dentate signal on both T1 and T2 sequences in four out of four subjects scanned. Brainstems were normal, apart from increased inferior olivary T2 signal in two, which is a neuroimaging correlate of symptomatic palatal tremor (Yokota et al., 1989). Illustrative scans are shown in Fig. 2. Reports on MRI scans in two other individuals, the films having now been destroyed, also refer to markedly decreased density in the region of the dentate nucleus, attributed to calcification.
Voice analysis was performed in the index case (III:11). The initial impression was of an ataxic dysarthria with spasmodic dysphonia. Stroboscopic laryngoscopy showed movement of laryngeal structures synchronously with the palatal tremor, with no evidence of adductor spasmodic dysphonia. Speech frequency analysis demonstrated abnormal pitch spread with pitch elevation, and voicing of non‐voiced sounds (Fig. 3). Nerve conduction studies (sural sensory, tibial motor) were performed in three, including one of those in whom the great toe two‐point discrimination threshold was >2 cm. All were normal. Calcium homeostasis was assessed (serum calcium, phosphate, magnesium, alkaline phosphatase, parathyroid hormone, 25‐hydroxy vitamin D) in five affected persons, with normal findings apart from a borderline low and borderline high level, respectively, of 25‐hydroxy vitamin D in IV:1 and IV:5, and borderline elevated parathyroid hormone levels in III:13 and IV:4, which we interpreted as being of no material significance. One individual (III:8) had a balanced chromosome translocation, 46,XY,t(1;12)(p22;q11), but this was not seen in several other family members karyotyped.
Genotyping and linkage analysis
SCAs 1, 2, 3, 6–8, 17 and DRPLA were excluded by direct CAG repeat expansion testing. Two‐point linkage analysis was performed with respect to SCAs 4–6 and 10–16, and no evidence of significant linkage was found (data not shown). A genome‐wide scan was undertaken. Single point analysis with MLINK showed a clear genome‐wide maximum LOD = 4.47, with a recombination fraction of 0, at marker D11S4191 (position 63.4 cM) in the pericentromeric region of chromosome 11 (Table 2). No other region tested significantly positive [thus also enabling exclusion of SCA18 (Brkanac et al., 2002), SCA19 (Verbeek et al., 2002), SCA21 (Vuillaume et al., 2002) and SCA22 (Chung et al., 2003)]. Analysis of the fine‐mapping data, using the VITESSE program, showed a clear peak at position 62.2 cM with a parametric LOD score of 4.51 (Fig. 4). (A 3‐LOD support is difficult to establish using the windowing approach, as an entire window spans the 3‐LOD support interval.)
The haplotype co‐segregating with the disease in this family is shown in Fig. 1. The informative recombination events observed in affected individuals III:13, IV:4 and IV:5 show that the distal recombination site is between markers D11S903 and D11S4109, and the recombination events in affected individuals IV:4 and IV:5 show that the proximal site is between markers D11S913 and FGF3. Using the genetic information from these individuals, the candidate region can be assigned to a 25.4 Mb interval between markers D11S903 and FGF3/INT2.
Triplet repeat analysis
No large CAG/CTG trinucleotide or ATTCT/AGAAT pentanucleotide repeats were identified.
Discussion
We describe a novel syndrome of cerebellar ataxia, dysphonia and palatal myoclonus. The rate of progression is slow and the degree of cerebellar affection is, for the most part, towards the less severe end of the SCA spectrum. There is no recognized instance of an affected person having an unaffected obligate heterozygous parent, thus indicating that the SCA20 gene is likely to be fully penetrant. The family data are insufficient formally to examine the case for anticipation. We have noted the datum of an average 10 year drop in (self‐reported) age of onset measured in three instances; against this, there is an apparent paucity of affected members in generation IV (possibly some are yet to manifest symptoms), and none is known to be affected in generation V. A further point against anticipation is that the RED analysis was negative, indicating that at least a large tri‐ or pentanucleotide expansion is unlikely to be the basis of the SCA20 mutation; nevertheless, we acknowledge that triplet repeat expansions exist in SCA8 (in the 3′‐untranslated region) (Koob et al., 1999) and in SCA12 (in the 5′‐untranslated region) (Holmes et al., 1999), and that neither of these SCAs shows anticipation.
The present syndrome may be distinguished clinically from the other SCAs by the associated features of dysphonia and palatal myoclonus, and the radiological observation of dentate calcification. Given this unique and rather striking combination of clinical features, we were expecting that our genetic analysis would identify a novel chromosomal region co‐segregating with the disease in the family. However, in fact, the syndrome proved to be linked to the same region of chromosome 11 within which the SCA5 locus lies. At first sight, a genetic identity between SCA5 and the present SCA might seem unlikely on phenotypic grounds, but that possibility has to remain open until such time as a genetic distinction might be proven (discussed further below). Nevertheless, a provisional assignment of the rubric SCA20 has been made by the Nomenclature Committee.
The abnormal dentate radiology is a very notable feature, and the development of the calcification appears to be an early feature in the evolution of the disease process, with one very subtly affected (IV:4) and two very mildly affected (III:12, III:16) persons having obvious signs on brain CT. Dentate calcification may be an incidental finding in 0.7% of those over 65 years of age, typically occurring in conjunction with pallidal calcification (Harrington et al., 1981). It is very rarely seen in the absence of basal ganglia calcification; in only one case from 4219 consecutive CT scans in the series of Koller et al. (1979). Hypoparathyroidism with basal ganglia calcification may be dominantly inherited (Smits et al., 1982), and dominant pseudohypoparathyroidism may cause a similar picture (Illum and Dupont, 1985; Flint and Goldstein, 1992). These conditions, for which otherwise there is no clinical support in our material, have been excluded on appropriate investigation. Microcalcification of the globus pallidus, but not the dentate, is recorded in Haw River syndrome, an unusual phenotypic variant of DRPLA (Farmer et al., 1989; Burke et al., 1994), scarcely a candidate clinical diagnosis in the present family (and excluded on both DNA and linkage analysis). Finally, a rare dominantly inherited disorder ‘familial idiopathic brain calcification’ has been described in some 13 pedigrees (Kobari et al., 1997; Prieto et al., 1997). This disorder may be asymptomatic despite radiological evidence of calcification; in those with clinical involvement, cognitive decline and parkinsonism have predominated over the cerebellar symptomatology. These additional clinical features were not present in SCA20; conversely, palatal tremor and dysphonia have not been described in familial idiopathic brain calcification. The distribution of calcification is also different in familial idiopathic brain calcification, with extensive involvement of basal ganglia, thalamus, cerebral cortex, subcortical white matter and hippocampus, in addition to dentate calcification. Finally, the condition maps, in at least one family, to a chromosome (number 14) different from SCA20 (Geschwind et al., 1999). We conclude that no other syndrome displays the same neuroradiological characteristics as the syndrome presently described.
Palatal tremor, sometimes also referred to as palatal or branchial myoclonus, may occur in isolated or symptomatic forms (Nagaoka and Narabayashi, 1984; Deuschl et al., 1990). SCA20 clearly falls into the symptomatic category, by reason of the associated clinical features, the absence of ear click and the spread of involvement to other branchial muscles (lips, pharynx) in three affected members. The abnormal inferior olivary signal on MRI in two of four members scanned (Fig. 2) is also consistent with the symptomatic form of palatal tremor, and probably represents olivary pseudohypertrophy (Yokota et al., 1989). The combination of palatal tremor and progressive ataxia can occur sporadically (Leger et al., 1986; Phanthumchinda, 1999), and it has been argued that it represents a unique degenerative syndrome (Sperling and Herrmann, 1985). It may be seen in the OPCA (olivopontocerebellar) variant of multiple system atrophy (Kulkarni et al., 1999). Palatal tremor with nystagmus, bulbar palsy and spastic tetraparesis has also been seen in autosomal dominant early adult‐onset Alexander’s disease (Howard et al., 1993; Schwankhaus et al., 1995; Okamoto et al., 2002), which is probably the same disorder as that described by de Yebenes et al. (1988) of a dominant branchial myoclonus with spastic paraparesis and cerebellar ataxia. While palatal tremor was consistently observed in these pedigrees, distinguishing them from SCAs 2 and 3 where it is an occasional feature only, no dentate calcification or dysphonia was evident. Later adult‐onset Alexander’s disease maps to chromosome 17 (Namekawa et al., 2002), although it is not yet clear whether all adult‐onset Alexander’s has the same genetic basis. Thus we conclude that, in all probability, on phenotypic and genetic grounds, these several families represent a different disorder from SCA20.
A single cerebellar ataxia disorder otherwise on record which includes dysphonia in the clinical picture can readily be separated from the present condition, with the dysphonia being due to a laryngeal abductor paralysis, and there being a concomitant motor neuropathy (Barbieri et al., 2001).
The classification of the dominantly inherited SCAs has shifted from a phenotypic towards a genotypic basis. The number of genetically defined SCA entities reported in the literature currently has reached 22 and will surely increase (Margolis, 2002). We need to consider the importance of SCA20 in the spectrum of the hereditary ataxias. The syndrome as described here is clinically distinctive enough that it would probably have been reported in the literature before now, were it a common cause of inherited ataxia. On the other hand, dentate calcification might well be viewed as incidental, and palatal tremor is occasionally recognized in other degenerative ataxias, so that a neurologist seeing a single member of a pedigree might well not appreciate that the disorder was novel (as was indeed the case with some in the present family). Ultimately, assessment of its rarity will have to await reports from others in other ethnic groups; the discovery that SCA12 is common in India although rare in the USA (where it was originally described) and the UK is an object lesson in this regard (Fujigasaki et al., 2001).
Our genetic analysis excluded all the cloned or mapped SCAs up to SCA22, with the exception of SCA5. An assumption of linkage to chromosome 11 can be made in light of the LOD score of >4. In the present family, informative recombinations in members III:13, IV:4 and IV:5 (Fig. 1) enabled narrowing down of the candidate region. One of the recombination sites is between markers D11S903 and D11S4109 and the other is between marker D11S913 and FGF3, and thus the candidate region extends from FGF3 to D11S903, this region having an estimated size of ∼25.4 Mb. The SCA5 candidate region, of size 5 cM, is bounded by FGF3 and PYGM, and this segment is wholly included within the SCA20 region. The exact ordering of markers in this region is uncertain, and clarification of the precise regions of overlap between the SCA5 and SCA20 candidate regions will be the subject of further study.
The question thus arises: is this an example of locus homogeneity, with SCAs 5 and 20 due to different mutations within the same gene? Or, are SCA5 and SCA20 genetically separate conditions, whose loci happen to lie quite closely together in the same chromosomal region? In terms of precedents for locus homogeneity, a number of neurological loci are known at which different types or sites of mutation may lead to different phenotypes. One example among the SCAs is the calcium channel gene CACNA1A, in which different mutations may lead to SCA6, hemiplegic migraine or episodic ataxia type 2 (Margolis, 2002); another potential example is the PRKCG locus, the basis of SCA14, this being typically a pure cerebellar ataxia, but in which some mutations may be associated with extracerebellar features (Van De Warrenburg et al., 2003; Yabe et al., 2003). Certainly, SCA5 differs clinically from SCA20. SCA5 was described initially by Ranum et al. (1994), and one further family was identified subsequently (Stevanin et al., 1999). The phenotype in the original family consisted of a pure cerebellar ataxia, with onset usually in the third or fourth decade, and a slow progression. In the second SCA5 family, the picture was similar but included a concomitant slight facial myokymia, increased reflexes and decreased vibration sensation. MRI studies in both families are reported as showing cerebellar atrophy without brainstem involvement, and in neither was any mention made of an abnormal dentate signal.
The alternative interpretation is that of a genetic heterogeneity between SCAs 5 and 20. The very distinctive phenotypic features of SCA20, as discussed above, might be seen as evidence against a genetic (locus) identity between the two. Along with the present potential case, two other SCA examples can be cited in which similar questions arise. SCA4 was reported in the original kindred with an accompanying axonal peripheral neuropathy, but a second ‘SCA4 family’ mapping to the same region displayed only a pure cerebellar ataxia (Nagaoka et al., 2000). We have described a pure cerebellar ataxia, SCA15, mapping to chromosome 3p26 (Knight et al., 2001; Storey et al., 2001), and a Japanese group since reported a SCA family with linkage to the same region, but in which there were the additional clinical features of tremor and myoclonus (Hara et al., 2002). However, distinction of the SCA5 and SCA20 genotypes can be made only in the knowledge of the actual causative gene(s) and, pending that knowledge, the issue of locus homogeneity versus coincidental (or possibly functional) locus propinquity remains open to question.
We conclude that the condition we describe is a clinically distinctive syndrome, and which may well prove to be a genetically distinct condition. If so, the assignment of a new SCA genetic rubric, SCA20, could in due course be confirmed. However, given its co‐localization to the same region of chromosome 11 which harbours the SCA5 locus, we concede that there exists the alternative explanation of phenotypic heterogeneity residing in SCA5. We intend to work to answer this question.
Addendum
After these studies had been undertaken, a further case was identified when the 40‐year‐old son of individual III:10 presented himself to our clinic. He showed mild dysarthria, slight dysphonia and subtle palatal myoclonus, and dentate calcification was seen on CT scan. Indices of calcium function (as above) were all normal.
Acknowledgements
We wish to thank the family members for their willing cooperation, Ms Janet H. Shaw for SCA gene analysis, and Mr Harry Rundle of the Alfred Hospital for performing the stroboscopic laryngoscopy. The study was supported by the Murdoch Childrens Research Institute, which is the recipient of a Centre Grant from the National Health and Medical Research Council of Australia. M.A.K. is a National Health and Medical Research Council Dora Lush Postgraduate Scholar. T.M. is supported by the National Ataxia Foundation, Minneapolis, USA, Ichiro Kanehara Foundation and Brain Science Foundation, Tokyo, Japan. The clinical material was presented as a poster at the 37th annual meeting of the Canadian Congress of Neurological Sciences, Vancouver, Canada, June 2002, and the genetic study was presented as a poster at the 52nd annual meeting of the American Society of Human Genetics, Baltimore, MD, October 2002.
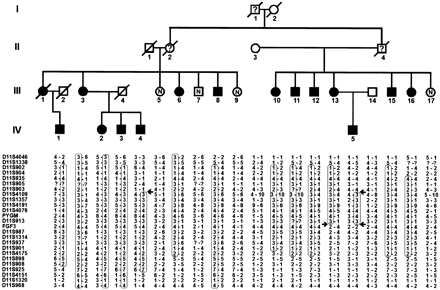
Fig. 1 Partial pedigree of the kindred and haplotyping results. In generation III, only those actually examined are included, and in generation IV, only those examined and diagnosed as affected, and it is upon these data that the linkage exercise is based (no definite judgement could be made about the genotypes of the unaffected individuals of generation IV, and they were therefore excluded). Haplotypes with respect to chromosome 11 markers have been constructed according to the most parsimonious requirement for recombination, the markers being ordered, from top to bottom, from p‐terminal to q‐terminal. The haplotype segregating with the disease is boxed, and arrows show limits of the shortest region of overlap, which thus defines the SCA20 candidate region, in the individuals in whom this could be inferred. The SCA5 region, which extends from FGF3 to PYGM, is included within this SCA20 region. Filled symbols = clinically affected on personal examination and/or dentate calcification on neuroradiology; N = no symptomatology, no signs of cerebellar disease on examination; ? = no reliable information (earlier generations); open symbols = unaffected spouse; diagonal line through symbol = deceased.
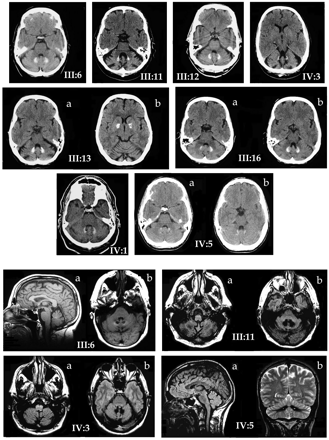
Fig. 2 Neuroradiology. Top three rows: non‐contrast axial brain CT scans on eight affected family members, showing dentate calcification in all, and pancerebellar atrophy in most. Cuts at two levels of the dentate are shown for III:16 and IV:5. Only one individual showed obvious pallidal calcification (III:13b); very slight pallidal calcification was seen in one other case (IV:3). Ages at examination range from 38 (IV:5) to 72 (III:6) years. Bottom two rows: non‐contrast brain MRI scans of four affected family members. III:6: T1 sagittal sequence (a) showing mild vermal atrophy with normal brainstem, and axial proton density image (b) showing low dentate signal. III:11, IV:3: axial proton density images showing inferior olivary hypertrophy (a), and low dentate signal (b). IV:5: proton density sagittal image (a) showing mild vermal atrophy and normal brainstem, and T2 coronal image showing low dentate signal and mild cerebellar atrophy (b).
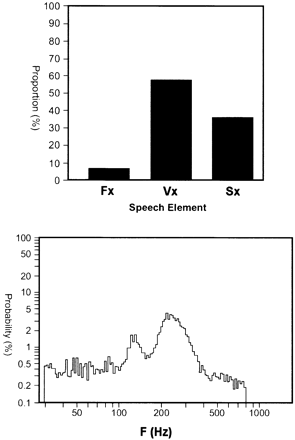
Fig. 3 Voice analysis in III:11. Top panel: proportion of frication (Fx), voiced (Vx) and intervocalic silence (Sx) time periods during passage reading, showing voicing of normally non‐voiced sounds (Vx and Sx are equal in normal subjects). Bottom panel: probability (log scale) versus frequency (F) (pitch) during passage reading. Normal (male) subjects produce a single, bell‐shaped curve, centred on average at ∼112 Hz. The frequency spread with multiple peaks and the high frequency of the maximum peak are abnormal in this subject.
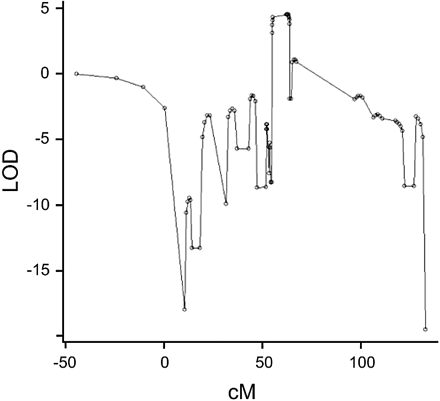
Fig. 4 Multipoint LOD score analysis with respect to fine‐mapping markers on chromosome 11. The maximum parametric LOD score achieved is 4.51 at position 62.2 cM.
Summary of certain clinical observations
| Case | Onset age/duration/age examined (years) | First symptom | ‘Dysphonia’ (see text) | Ballistic overshoot | Bradykinesia* | Tremor |
| III:3 | 57, 20, 77 | Dysarthria | Mild to moderate | Moderate | Bilaterally ↓ 2.5 SD | Palatal |
| III:6 | 64, 10, 74 | Gait ataxia | Mild | Moderate | Bilaterally ↑ 1.5 SD | Nil |
| III:8 | 45, 20, 65 | Dysarthria and ataxia | Nil | Moderate right, marked left | Right ↓ 1 SD, left ↓ 2 SD | Palatal, lower lip |
| III:10 | 60, 8, 68 | Dysarthria | Mild | Moderate | Right ↑ 1.5 SD, left ↑ 0.5 SD | Palatal |
| III:11 | 48, 20, 67 | Dysarthria | Moderate | Moderate | Bilaterally ↓ 1 SD | Nil |
| III:12 | 62, 3, 65 | Dysarthria and ataxia | Very mild | Mild right, moderate left | Right ↑ 1 SD, left average | Palatal |
| III:13 | 40, 24, 64 | Dysarthria | Moderate | Mild to moderate | Bilaterally ↑ 0.5 SD | Palatal, upper lip |
| III:15 | 19, 43, 62 | Dysarthria | Moderate | Marked | Bilaterally ↓ 3 SD | Palatal |
| III:16 | 56, 1, 56 | Dysarthria | Very mild | Nil right, mild left | Right ↑ 1 SD, left average | Palatal |
| IV:1 | 55, 3, 58 | Gait ataxia | Nil | Marked right, moderate left | Bilaterally ↓ 3.5 SD | Palatal |
| IV:2 | 42, 15, 57 | Dysarthria | Nil | Mild | Bilaterally ↓ 2.5 SD | Palatal |
| IV:3 | 38, 16, 54 | Dysarthria (sudden onset) | Mild | Marked | Bilaterally ↓ 2 SD | Palatal, head, arm |
| IV:4** | 33, 5, 38 | Tremor | Nil | Mild right, nil left | Not tested | Nil |
| IV:5 | 34, 5, 39 | Dysarthria (sudden onset) | Very mild | Moderate right, mild left | Bilaterally ↓ 3 SD | Nil |
| Case | Onset age/duration/age examined (years) | First symptom | ‘Dysphonia’ (see text) | Ballistic overshoot | Bradykinesia* | Tremor |
| III:3 | 57, 20, 77 | Dysarthria | Mild to moderate | Moderate | Bilaterally ↓ 2.5 SD | Palatal |
| III:6 | 64, 10, 74 | Gait ataxia | Mild | Moderate | Bilaterally ↑ 1.5 SD | Nil |
| III:8 | 45, 20, 65 | Dysarthria and ataxia | Nil | Moderate right, marked left | Right ↓ 1 SD, left ↓ 2 SD | Palatal, lower lip |
| III:10 | 60, 8, 68 | Dysarthria | Mild | Moderate | Right ↑ 1.5 SD, left ↑ 0.5 SD | Palatal |
| III:11 | 48, 20, 67 | Dysarthria | Moderate | Moderate | Bilaterally ↓ 1 SD | Nil |
| III:12 | 62, 3, 65 | Dysarthria and ataxia | Very mild | Mild right, moderate left | Right ↑ 1 SD, left average | Palatal |
| III:13 | 40, 24, 64 | Dysarthria | Moderate | Mild to moderate | Bilaterally ↑ 0.5 SD | Palatal, upper lip |
| III:15 | 19, 43, 62 | Dysarthria | Moderate | Marked | Bilaterally ↓ 3 SD | Palatal |
| III:16 | 56, 1, 56 | Dysarthria | Very mild | Nil right, mild left | Right ↑ 1 SD, left average | Palatal |
| IV:1 | 55, 3, 58 | Gait ataxia | Nil | Marked right, moderate left | Bilaterally ↓ 3.5 SD | Palatal |
| IV:2 | 42, 15, 57 | Dysarthria | Nil | Mild | Bilaterally ↓ 2.5 SD | Palatal |
| IV:3 | 38, 16, 54 | Dysarthria (sudden onset) | Mild | Marked | Bilaterally ↓ 2 SD | Palatal, head, arm |
| IV:4** | 33, 5, 38 | Tremor | Nil | Mild right, nil left | Not tested | Nil |
| IV:5 | 34, 5, 39 | Dysarthria (sudden onset) | Very mild | Moderate right, mild left | Bilaterally ↓ 3 SD | Nil |
*Index tapping rate per 10 s (Bornstein, 1985); **affected status confirmed on neuroradiology (dentate calcification on CT scan).
Summary of certain clinical observations
| Case | Onset age/duration/age examined (years) | First symptom | ‘Dysphonia’ (see text) | Ballistic overshoot | Bradykinesia* | Tremor |
| III:3 | 57, 20, 77 | Dysarthria | Mild to moderate | Moderate | Bilaterally ↓ 2.5 SD | Palatal |
| III:6 | 64, 10, 74 | Gait ataxia | Mild | Moderate | Bilaterally ↑ 1.5 SD | Nil |
| III:8 | 45, 20, 65 | Dysarthria and ataxia | Nil | Moderate right, marked left | Right ↓ 1 SD, left ↓ 2 SD | Palatal, lower lip |
| III:10 | 60, 8, 68 | Dysarthria | Mild | Moderate | Right ↑ 1.5 SD, left ↑ 0.5 SD | Palatal |
| III:11 | 48, 20, 67 | Dysarthria | Moderate | Moderate | Bilaterally ↓ 1 SD | Nil |
| III:12 | 62, 3, 65 | Dysarthria and ataxia | Very mild | Mild right, moderate left | Right ↑ 1 SD, left average | Palatal |
| III:13 | 40, 24, 64 | Dysarthria | Moderate | Mild to moderate | Bilaterally ↑ 0.5 SD | Palatal, upper lip |
| III:15 | 19, 43, 62 | Dysarthria | Moderate | Marked | Bilaterally ↓ 3 SD | Palatal |
| III:16 | 56, 1, 56 | Dysarthria | Very mild | Nil right, mild left | Right ↑ 1 SD, left average | Palatal |
| IV:1 | 55, 3, 58 | Gait ataxia | Nil | Marked right, moderate left | Bilaterally ↓ 3.5 SD | Palatal |
| IV:2 | 42, 15, 57 | Dysarthria | Nil | Mild | Bilaterally ↓ 2.5 SD | Palatal |
| IV:3 | 38, 16, 54 | Dysarthria (sudden onset) | Mild | Marked | Bilaterally ↓ 2 SD | Palatal, head, arm |
| IV:4** | 33, 5, 38 | Tremor | Nil | Mild right, nil left | Not tested | Nil |
| IV:5 | 34, 5, 39 | Dysarthria (sudden onset) | Very mild | Moderate right, mild left | Bilaterally ↓ 3 SD | Nil |
| Case | Onset age/duration/age examined (years) | First symptom | ‘Dysphonia’ (see text) | Ballistic overshoot | Bradykinesia* | Tremor |
| III:3 | 57, 20, 77 | Dysarthria | Mild to moderate | Moderate | Bilaterally ↓ 2.5 SD | Palatal |
| III:6 | 64, 10, 74 | Gait ataxia | Mild | Moderate | Bilaterally ↑ 1.5 SD | Nil |
| III:8 | 45, 20, 65 | Dysarthria and ataxia | Nil | Moderate right, marked left | Right ↓ 1 SD, left ↓ 2 SD | Palatal, lower lip |
| III:10 | 60, 8, 68 | Dysarthria | Mild | Moderate | Right ↑ 1.5 SD, left ↑ 0.5 SD | Palatal |
| III:11 | 48, 20, 67 | Dysarthria | Moderate | Moderate | Bilaterally ↓ 1 SD | Nil |
| III:12 | 62, 3, 65 | Dysarthria and ataxia | Very mild | Mild right, moderate left | Right ↑ 1 SD, left average | Palatal |
| III:13 | 40, 24, 64 | Dysarthria | Moderate | Mild to moderate | Bilaterally ↑ 0.5 SD | Palatal, upper lip |
| III:15 | 19, 43, 62 | Dysarthria | Moderate | Marked | Bilaterally ↓ 3 SD | Palatal |
| III:16 | 56, 1, 56 | Dysarthria | Very mild | Nil right, mild left | Right ↑ 1 SD, left average | Palatal |
| IV:1 | 55, 3, 58 | Gait ataxia | Nil | Marked right, moderate left | Bilaterally ↓ 3.5 SD | Palatal |
| IV:2 | 42, 15, 57 | Dysarthria | Nil | Mild | Bilaterally ↓ 2.5 SD | Palatal |
| IV:3 | 38, 16, 54 | Dysarthria (sudden onset) | Mild | Marked | Bilaterally ↓ 2 SD | Palatal, head, arm |
| IV:4** | 33, 5, 38 | Tremor | Nil | Mild right, nil left | Not tested | Nil |
| IV:5 | 34, 5, 39 | Dysarthria (sudden onset) | Very mild | Moderate right, mild left | Bilaterally ↓ 3 SD | Nil |
*Index tapping rate per 10 s (Bornstein, 1985); **affected status confirmed on neuroradiology (dentate calcification on CT scan).
Two‐point LOD scores for the SCA20 locus and 24 chromosome 11 markers
| Genome‐wide scan markers | Fine‐mapping markers | Location (cM) | Recombination fraction (θ) | ||||||
| 0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | |||
| D11S4046 | –18.11 | –5.89 | –2.58 | –1.32 | –0.34 | –0.02 | 0.02 | ||
| D11S1338 | –13.06 | –4.4 | –1.79 | –0.82 | –0.12 | 0.06 | 0.04 | ||
| D11S902 | 24.7 | –9.82 | –1.99 | –0.74 | –0.31 | –0.02 | 0.06 | 0.06 | |
| D11S904 | 37.0 | –3.18 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | |
| D11S935 | 49.6 | –8.73 | –0.23 | 0.93 | 1.23 | 1.18 | 0.84 | 0.36 | |
| D11S905 | 55.7 | –2.7 | 1.47 | 1.94 | 1.95 | 1.61 | 1.1 | 0.47 | |
| D11S903 | 59.5 | –9.06 | –0.47 | 0.71 | 1.04 | 1.05 | 0.75 | 0.31 | |
| D11S1357 | 62.5 | 3.53 | 3.47 | 3.2 | 2.86 | 2.15 | 1.39 | 0.6 | |
| D11S4109 | 63.3 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4191 | 63.4 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4076 | 64.9 | 4.17 | 4.1 | 3.81 | 3.43 | 2.63 | 1.76 | 0.8 | |
| PYGM | 2.05 | 2.01 | 1.82 | 1.61 | 1.19 | 0.8 | 0.41 | ||
| D11S913 | 70.9 | 3.04 | 2.99 | 2.75 | 2.46 | 1.83 | 1.18 | 0.51 | |
| INT2 (FGF3) | –3.23 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | ||
| D11S987 | –1.95 | 2.38 | 2.8 | 2.73 | 2.25 | 1.55 | 0.72 | ||
| D11S1314 | 77.5 | –3 | 1 | 1.46 | 1.46 | 1.16 | 0.73 | 0.28 | |
| D11S937 | 84.6 | –1.93 | 2.39 | 2.81 | 2.75 | 2.26 | 1.57 | 0.73 | |
| D11S901 | 89.8 | –2.77 | 0.99 | 1.46 | 1.46 | 1.14 | 0.68 | 0.23 | |
| D11S4175 | –3.36 | –1.01 | 0.2 | 0.55 | 0.64 | 0.46 | 0.19 | ||
| D11S908 | –3.39 | –2.85 | –1.07 | –0.38 | 0.07 | 0.14 | 0.07 | ||
| D11S925 | –8.65 | –4.29 | –2.78 | –1.52 | –0.45 | –0.03 | 0.08 | ||
| D11S4151 | –19.55 | –9.36 | –5.15 | –3.24 | –1.51 | –0.66 | –0.2 | ||
| D11S1320 | –9.11 | –6.57 | –3.66 | –2.29 | –1.09 | –0.52 | –0.19 | ||
| D11S968 | 0.66 | 0.65 | 0.57 | 0.47 | 0.29 | 0.14 | 0.05 | ||
| Genome‐wide scan markers | Fine‐mapping markers | Location (cM) | Recombination fraction (θ) | ||||||
| 0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | |||
| D11S4046 | –18.11 | –5.89 | –2.58 | –1.32 | –0.34 | –0.02 | 0.02 | ||
| D11S1338 | –13.06 | –4.4 | –1.79 | –0.82 | –0.12 | 0.06 | 0.04 | ||
| D11S902 | 24.7 | –9.82 | –1.99 | –0.74 | –0.31 | –0.02 | 0.06 | 0.06 | |
| D11S904 | 37.0 | –3.18 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | |
| D11S935 | 49.6 | –8.73 | –0.23 | 0.93 | 1.23 | 1.18 | 0.84 | 0.36 | |
| D11S905 | 55.7 | –2.7 | 1.47 | 1.94 | 1.95 | 1.61 | 1.1 | 0.47 | |
| D11S903 | 59.5 | –9.06 | –0.47 | 0.71 | 1.04 | 1.05 | 0.75 | 0.31 | |
| D11S1357 | 62.5 | 3.53 | 3.47 | 3.2 | 2.86 | 2.15 | 1.39 | 0.6 | |
| D11S4109 | 63.3 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4191 | 63.4 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4076 | 64.9 | 4.17 | 4.1 | 3.81 | 3.43 | 2.63 | 1.76 | 0.8 | |
| PYGM | 2.05 | 2.01 | 1.82 | 1.61 | 1.19 | 0.8 | 0.41 | ||
| D11S913 | 70.9 | 3.04 | 2.99 | 2.75 | 2.46 | 1.83 | 1.18 | 0.51 | |
| INT2 (FGF3) | –3.23 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | ||
| D11S987 | –1.95 | 2.38 | 2.8 | 2.73 | 2.25 | 1.55 | 0.72 | ||
| D11S1314 | 77.5 | –3 | 1 | 1.46 | 1.46 | 1.16 | 0.73 | 0.28 | |
| D11S937 | 84.6 | –1.93 | 2.39 | 2.81 | 2.75 | 2.26 | 1.57 | 0.73 | |
| D11S901 | 89.8 | –2.77 | 0.99 | 1.46 | 1.46 | 1.14 | 0.68 | 0.23 | |
| D11S4175 | –3.36 | –1.01 | 0.2 | 0.55 | 0.64 | 0.46 | 0.19 | ||
| D11S908 | –3.39 | –2.85 | –1.07 | –0.38 | 0.07 | 0.14 | 0.07 | ||
| D11S925 | –8.65 | –4.29 | –2.78 | –1.52 | –0.45 | –0.03 | 0.08 | ||
| D11S4151 | –19.55 | –9.36 | –5.15 | –3.24 | –1.51 | –0.66 | –0.2 | ||
| D11S1320 | –9.11 | –6.57 | –3.66 | –2.29 | –1.09 | –0.52 | –0.19 | ||
| D11S968 | 0.66 | 0.65 | 0.57 | 0.47 | 0.29 | 0.14 | 0.05 | ||
Two‐point LOD scores for the SCA20 locus and 24 chromosome 11 markers
| Genome‐wide scan markers | Fine‐mapping markers | Location (cM) | Recombination fraction (θ) | ||||||
| 0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | |||
| D11S4046 | –18.11 | –5.89 | –2.58 | –1.32 | –0.34 | –0.02 | 0.02 | ||
| D11S1338 | –13.06 | –4.4 | –1.79 | –0.82 | –0.12 | 0.06 | 0.04 | ||
| D11S902 | 24.7 | –9.82 | –1.99 | –0.74 | –0.31 | –0.02 | 0.06 | 0.06 | |
| D11S904 | 37.0 | –3.18 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | |
| D11S935 | 49.6 | –8.73 | –0.23 | 0.93 | 1.23 | 1.18 | 0.84 | 0.36 | |
| D11S905 | 55.7 | –2.7 | 1.47 | 1.94 | 1.95 | 1.61 | 1.1 | 0.47 | |
| D11S903 | 59.5 | –9.06 | –0.47 | 0.71 | 1.04 | 1.05 | 0.75 | 0.31 | |
| D11S1357 | 62.5 | 3.53 | 3.47 | 3.2 | 2.86 | 2.15 | 1.39 | 0.6 | |
| D11S4109 | 63.3 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4191 | 63.4 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4076 | 64.9 | 4.17 | 4.1 | 3.81 | 3.43 | 2.63 | 1.76 | 0.8 | |
| PYGM | 2.05 | 2.01 | 1.82 | 1.61 | 1.19 | 0.8 | 0.41 | ||
| D11S913 | 70.9 | 3.04 | 2.99 | 2.75 | 2.46 | 1.83 | 1.18 | 0.51 | |
| INT2 (FGF3) | –3.23 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | ||
| D11S987 | –1.95 | 2.38 | 2.8 | 2.73 | 2.25 | 1.55 | 0.72 | ||
| D11S1314 | 77.5 | –3 | 1 | 1.46 | 1.46 | 1.16 | 0.73 | 0.28 | |
| D11S937 | 84.6 | –1.93 | 2.39 | 2.81 | 2.75 | 2.26 | 1.57 | 0.73 | |
| D11S901 | 89.8 | –2.77 | 0.99 | 1.46 | 1.46 | 1.14 | 0.68 | 0.23 | |
| D11S4175 | –3.36 | –1.01 | 0.2 | 0.55 | 0.64 | 0.46 | 0.19 | ||
| D11S908 | –3.39 | –2.85 | –1.07 | –0.38 | 0.07 | 0.14 | 0.07 | ||
| D11S925 | –8.65 | –4.29 | –2.78 | –1.52 | –0.45 | –0.03 | 0.08 | ||
| D11S4151 | –19.55 | –9.36 | –5.15 | –3.24 | –1.51 | –0.66 | –0.2 | ||
| D11S1320 | –9.11 | –6.57 | –3.66 | –2.29 | –1.09 | –0.52 | –0.19 | ||
| D11S968 | 0.66 | 0.65 | 0.57 | 0.47 | 0.29 | 0.14 | 0.05 | ||
| Genome‐wide scan markers | Fine‐mapping markers | Location (cM) | Recombination fraction (θ) | ||||||
| 0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | |||
| D11S4046 | –18.11 | –5.89 | –2.58 | –1.32 | –0.34 | –0.02 | 0.02 | ||
| D11S1338 | –13.06 | –4.4 | –1.79 | –0.82 | –0.12 | 0.06 | 0.04 | ||
| D11S902 | 24.7 | –9.82 | –1.99 | –0.74 | –0.31 | –0.02 | 0.06 | 0.06 | |
| D11S904 | 37.0 | –3.18 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | |
| D11S935 | 49.6 | –8.73 | –0.23 | 0.93 | 1.23 | 1.18 | 0.84 | 0.36 | |
| D11S905 | 55.7 | –2.7 | 1.47 | 1.94 | 1.95 | 1.61 | 1.1 | 0.47 | |
| D11S903 | 59.5 | –9.06 | –0.47 | 0.71 | 1.04 | 1.05 | 0.75 | 0.31 | |
| D11S1357 | 62.5 | 3.53 | 3.47 | 3.2 | 2.86 | 2.15 | 1.39 | 0.6 | |
| D11S4109 | 63.3 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4191 | 63.4 | 4.47 | 4.4 | 4.1 | 3.71 | 2.87 | 1.94 | 0.91 | |
| D11S4076 | 64.9 | 4.17 | 4.1 | 3.81 | 3.43 | 2.63 | 1.76 | 0.8 | |
| PYGM | 2.05 | 2.01 | 1.82 | 1.61 | 1.19 | 0.8 | 0.41 | ||
| D11S913 | 70.9 | 3.04 | 2.99 | 2.75 | 2.46 | 1.83 | 1.18 | 0.51 | |
| INT2 (FGF3) | –3.23 | 0.74 | 1.26 | 1.32 | 1.12 | 0.75 | 0.3 | ||
| D11S987 | –1.95 | 2.38 | 2.8 | 2.73 | 2.25 | 1.55 | 0.72 | ||
| D11S1314 | 77.5 | –3 | 1 | 1.46 | 1.46 | 1.16 | 0.73 | 0.28 | |
| D11S937 | 84.6 | –1.93 | 2.39 | 2.81 | 2.75 | 2.26 | 1.57 | 0.73 | |
| D11S901 | 89.8 | –2.77 | 0.99 | 1.46 | 1.46 | 1.14 | 0.68 | 0.23 | |
| D11S4175 | –3.36 | –1.01 | 0.2 | 0.55 | 0.64 | 0.46 | 0.19 | ||
| D11S908 | –3.39 | –2.85 | –1.07 | –0.38 | 0.07 | 0.14 | 0.07 | ||
| D11S925 | –8.65 | –4.29 | –2.78 | –1.52 | –0.45 | –0.03 | 0.08 | ||
| D11S4151 | –19.55 | –9.36 | –5.15 | –3.24 | –1.51 | –0.66 | –0.2 | ||
| D11S1320 | –9.11 | –6.57 | –3.66 | –2.29 | –1.09 | –0.52 | –0.19 | ||
| D11S968 | 0.66 | 0.65 | 0.57 | 0.47 | 0.29 | 0.14 | 0.05 | ||
References
Barbieri F, Pellecchia MT, Esposito E, Di Stasio E, Castaldo I, Santorelli F, et al. Adult‐onset familial laryngeal abductor paralysis, cerebellar ataxia, and pure motor neuropathy.
Bornstein RA. Normative data on seleted neuropsychological measures from a nonclinical sample.
Brkanac Z, Fernandez M, Matsushita M, Lipe H, Wolff J, Bird TD, et al. Autosomal dominant sensory/motor neuropathy with ataxia (SMNA): linkage to chromosome 7q22–q32.
Burke JR, Wingfield MS, Lewis KE, Roses AD, Lee JE, Hulette C, et al. The Haw River syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African‐American family.
Chung MY, Lu YC, Cheng NC, Soong BW. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21–q23.
de Yebenes JG, Vazquez A, Rabano J, de Seijas EV, Urra DG, Obregon MC, et al. Hereditary branchial myoclonus with spastic paraparesis and cerebellar ataxia: a new autosomal dominant disorder.
Deuschl G, Mischke G, Sohenck E, Schulte‐Monting J, Lucking CH. Symptomatic and essential rhythmic palatal myoclonus.
Farmer TW, Wingfield MS, Lynch SA, Vogel FS, Hulette C, Katchinoff B, et al. Ataxia, chorea, seizures, and dementia. Pathologic features of a newly defined familial disorder.
Flanigan K, Gardner K, Alderson K, Galster B, Otterud B, Leppert MF, et al. Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): clinical description and genetic localization to chromosome 16q22.1.
Flint J, Goldstein LH. Familial calcification of the basal ganglia: a case report and review of the literature.
Fujigasaki H, Verma IC, Camuzat A, Margolis RL, Zander C, Lebre AS, et al. SCA12 is a rare locus for autosomal dominant cerebellar ataxia: a study of an Indian family.
Geschwind DH, Loginov M, Stern JM. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification (Fahr disease).
Hara K, Fukusihima T, Shimohata T, Oyake M, Ishiguro H, Hirota K, et al. A novel autosomal dominant cerebellar ataxia linked to chromosome 3p26 [abstract].
Harrington MG, Macpherson P, McIntosh WB, Allam BF, Bone I. The significance of the incidental finding of basal ganglia calcification on computed tomography.
Herman‐Bert A, Stevanin G, Netter JC, Rascol O, Brassat D, Calvas P, et al. Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3–q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation.
Holmes SE, O’Hearn EE, McInnis MG, Gorelick‐Feldman DA, Kleiderlein JJ, Callahan C, et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12 [letter].
Howard RS, Greenwood R, Gawler J, Scaravilli F, Marsden CD, Harding AE. A familial disorder associated with palatal myoclonus, other brainstem signs, tetraparesis, ataxia and Rosenthal fibre formation.
Illum F, Dupont E. Prevalences of CT‐detected calcification in the basal ganglia in idiopathic hypoparathyroidism and pseudohypoparathyroidism.
Knight MA, Kennerson ML, Nicholson GA, Gardner RJM, Storey E, Thomas PQ, et al. A new spinocerebellar ataxia, SCA15.
Knight MA, Kennerson ML, Anney RJ, Matsuura T, Nicholson GA, Salimi‐Tari P, et al. Spinocerebellar ataxia type 15 (SCA15) maps to 3p24.2–3pter: exclusion of the ITPR1 gene, the human orthologue of an ataxic mouse mutant.
Kobari M, Nogawa S, Sugimoto Y, Fukuuchi Y. Familial idiopathic brain calcification with autosomal dominant inheritance.
Koller WC, Cochran JW, Klawans HL. Calcification of the basal ganglia: computerized tomography and clinical correlation.
Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8).
Kulkarni PK, Muthane UB, Taly AB, Jayakumar PN, Shetty R, Swamy HS. Palatal tremor, progressive multiple cranial nerve palsies, and cerebellar ataxia: a case report and review of literature of palatal tremors in neurodegenerative disease.
Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkage analysis in humans.
Leger JM, Duyckaerts C, Brunet P. Syndrome of palatal myoclonus and progressive ataxia: report of a case.
Margolis RL. The spinocerebellar ataxias: order emerges from chaos.
Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10.
McPeek MS, Sun L. Statistical tests for detection of misspecified relationships by use of genome‐screen data.
Miyoshi Y, Yamada T, Tanimura M, Taniwaki T, Arakawa K, Ohyagi Y, et al. A novel autosomal dominant spinocerebellar ataxia (SCA16) linked to chromosome 8q22.1–24.1.
Nagaoka M, Narabayashi H. Palatal myoclonus—its remote influence.
Nagaoka U, Takashima M, Ishikawa K, Yoshizawa K, Yoshizawa T, Ishikawa M, et al. A gene on SCA4 locus causes dominantly inherited pure cerebellar ataxia.
Namekawa M, Takiyama Y, Aoki Y, Takayashiki N, Sakoe K, Shimazaki H, et al. Identification of GFAP gene mutation in hereditary adult‐onset Alexander’s disease.
O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis.
Okamoto Y, Mitsuyama H, Jonosono M, Hirata K, Arimura K, Osame M, et al. Autosomal dominant palatal myoclonus and spinal cord atrophy.
Phanthumchinda K. Syndrome of progressive ataxia and palatal myoclonus: a case report.
Ploughman LM, Boehnke M. Estimating the power of a proposed linkage study for a complex genetic trait.
Prieto JM, Pardellas H, Sobrido MJ, Lema M, Dapena D, Castro A. Calcificación estriopalidodentada familiar.
Ranum LP, Schut LJ, Lundgren JK, Orr HT, Livingston DM. Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11.
Schalling M, Hudson TJ, Buetow KH, Housman DE. Direct detection of novel expanded trinucleotide repeats in the human genome.
Schwankhaus JD, Parisi JE, Gulledge WR, Chin L, Currier RD. Hereditary adult‐onset Alexander’s disease with palatal myoclonus, spastic paraparesis, and cerebellar ataxia.
Smits M, Gabreels F, Froeling P, Thijssen H, Colon E, ter Haar B, et al. Autosomal dominant idiopathic hypoparathyroidism and nervous system dysfunction: report of three cases and review of the literature.
Sperling MR, Herrmann C. Syndrome of palatal myoclonus and progressive ataxia: two cases with magnetic resonance imaging.
Stevanin G, Herman A, Brice A, Durr A. Clinical and MRI findings in spinocerebellar ataxia type 5.
Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, Gardner RJM, et al. Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia.
Storey E, Gardner RJM, Knight MA, Kennerson ML, Tuck RR, Forrest SM, et al. A new autosomal dominant pure cerebellar ataxia.
Subramony SH, Filla A. Autosomal dominant spinocerebellar ataxias ad infinitum?
Terwilliger JD, Ott J. Handbook of human genetic linkage. Baltimore: Johns Hopkins University Press;
Van De Warrenburg BP, Verbeek DS, Piersma SJ, Hennekam FA, Pearson PL, Knoers NV, et al. Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family.
Verbeek DS, Schelhaas JH, Ippel EF, Beemer FA, Pearson PL, Sinke RJ. Identification of a novel SCA locus (SCA19) in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21–q21.
Vuillaume I, Devos D, Schraen‐Maschke S, Dina C, Lemainque A, Vasseur F, et al. A new locus for spinocerebellar ataxia (SCA21) maps to chromosome 7p21.3–p15.1.
Worth PF, Giunti P, Gardner‐Thorpe C, Dixon PH, Davis MB, Wood NW. Autosomal dominant cerebellar ataxia type III: linkage in a large British family to a 7.6‐cM region on chromosome 15q14–21.3.
Yabe I, Sasaki H, Chen DH, Raskind WH, Bird TD, Yamashita I, et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma.
Yamashita I, Sasaki H, Yabe I, Fukazawa T, Nogoshi S, Komeichi K, et al. A novel locus for dominant cerebellar ataxia (SCA14) maps to a 10.2‐cM interval flanked by D19S206 and D19S605 on chromosome 19q13.4–qter.
Yokota T, Hirashima F, Furukawa T, Tsukagoshi H, Yoshikawa H. MRI findings of inferior olives in palatal myoclonus.
Yue Q, Jen JC, Nelson SF, Baloh RW. Progressive ataxia due to a missense mutation in a calcium‐channel gene.
Zee DS. Ophthalmoscopy in examination of patients with vestibular disorders.


{kind=link}
{kind=link}
{kind=link}
{kind=link}