




Abstract
Mutations in the SPG7 gene, encoding the mitochondrial protein paraplegin, were the first to be identified in autosomal recessive hereditary spastic paraplegia (ARHSP). Four different SPG7 mutations have been described so far in association with both pure and complicated HSP phenotypes. Muscle biopsies from the most severely affected patients have shown histological evidence of an oxidative phosphorylation defect. We identified six ARHSP kindreds, in whom linkage to SPG7 could not be excluded, and 29 sporadic spastic paraplegia patients. The 17 exons and flanking regions of the SPG7 gene were screened for mutations using a combination of single‐stranded conformation polymorphism (SSCP) analysis and sequencing. Three patients were found to carry compound heterozygous SPG7 mutations, comprising five novel and one previously described mutation. Muscle biopsies from two SPG7 mutation patients did not show any histological evidence of an oxidative phosphorylation defect. However, biochemical analysis revealed a reduction in citrate synthase‐corrected complex I and complex II/III activities in muscle and complex I activity in mitochondrial‐enriched fractions from cultured myoblasts, suggesting that either a primary or a secondary defect of respiratory chain function may play an important role in the pathogenesis of the disease.
Introduction
Hereditary spastic paraplegia (HSP) encompasses a group of clinically and genetically heterogeneous disorders in which the predominant feature is progressive spasticity and weakness of the lower limbs. Inheritance may be autosomal dominant, autosomal recessive (ARHSP) or X‐linked. The phenotype is classified as pure HSP when symptoms and signs are confined to those of a progressive spastic paraparesis with possible posterior column or bladder involvement (Polo et al., 1993). Complicated HSP phenotypes have been described in association with a wide range of additional neurological or other clinical features (McDermott and Shaw, 2002).
The majority of HSP cases (∼80%) demonstrate evidence of autosomal dominant inheritance (Polo et al., 1991). To date, 10 autosomal dominant HSP loci have been mapped, from which mutations in five genes, SPG3A (atlastin), SPG4 (spastin), SPG6 (NIPA1), SPG10 (KIF5A) and SPG13 (Hsp60), have been identified (Hazan et al., 1999; Zhao et al., 2001; Hansen et al., 2002; Reid et al., 2002; Rainier et al., 2003). Autosomal recessive inheritance appears relatively uncommon outside regions where there are high rates of consanguineous marriages (Sridharan et al., 1985), although they may comprise a significant proportion of apparently sporadic cases. To date, six ARHSP loci have been identified and causative mutations found in two genes; SPG7, encoding paraplegin, and SPG20, encoding the protein spartin (Casari et al., 1998; Patel et al., 2002).
SPG7 was the first autosomal HSP gene to be characterized. Located on chromosome 16, it comprises 17 exons spanning ∼52 kb. The protein product, paraplegin, consists of 795 amino acids and localizes to mitochondria. It shares its closest amino acid sequence homology with the yeast mitochondrial metalloproteases Afg3, Rca1 and Yme1 (Casari et al., 1998; Settasatian et al., 1999). These proteins are members of the AAA protein superfamily (ATPase associated with diverse cellular activities) which are found widely in both prokaryotic and eukaryotic cells and play an important role in a variety of cellular activities including cell division, transcription, organelle biogenesis, vesicle transport and enzyme assembly (Patel and Latterich, 1998). Yeast mitochondrial ATPases possess both proteolytic and chaperone‐like activities at the inner mitochondrial membrane where they are involved in the assembly and degradation of proteins in the respiratory chain complex (Pearce, 1999). Mutation of these genes in yeast induces a respiratory chain defect due to a block in the assembly of the subunits into functional enzymes (Paul and Tzagoloff, 1995). Subsequently, a number of additional human genes encoding proteins highly homologous to paraplegin such as AFG3L1, AFG3L2 and YME1L1 have been discovered (Banfi et al., 1999; Coppola et al., 2000; Kremmidiotis et al., 2001). The precise function of paraplegin and related proteins in humans remains uncertain.
To date, four SPG7 mutations have been described resulting in either pure or complicated HSP phenotypes. Following linkage to 16q24.3 in a large consanguineous Italian family with a variable complicated phenotype (De Michele et al., 1998), Casari et al. (1998) discovered a 9.5 kb deletion corresponding to the last five exons of the SPG7 gene. Muscle biopsy analysis from two severely affected individuals revealed characteristic changes of mitochondrial oxidative phosphorylation defects including ragged‐red fibres, cytochrome c oxidase‐negative and succinate dehydrogenase‐positive fibres. Electron microscopy confirmed an accumulation of abnormal mitochondria containing paracrystalline inclusions in a ‘parking lot’ pattern. However, in two less severely affected individuals, only a few scattered cytochrome c oxidase‐negative fibres were seen. Two additional frameshift mutations, a 2 bp deletion in exon 6 in a small Italian family with pure HSP and a single base insertion in exon 17 in a French HSP kindred with a complicated phenotype including optic, cortical and cerebellar atrophy, were also identified. Both families were consanguineous and homozygous for the mutation (Casari et al., 1998). Recently, a heterozygous 9 bp deletion in exon 11 of SPG7 has been reported in a father and son affected by a spastic paraparesis, suggesting a possible autosomal dominant mode of inheritance in this family (McDermott et al., 2001).
The aim of this study was to screen a large cohort of autosomal recessive and sporadic HSP cases from the UK to estimate the prevalence of SPG7 mutations in this population and further characterize the nature of mitochondrial dysfunction in affected individuals.
Methods
Subjects
Twenty HSP kindreds with evidence of autosomal recessive inheritance and a further 29 sporadic patients with no family history, in whom alternative causes for a spastic paraplegia had been excluded, were identified either through an established database of British HSP patients (based at the three participating centres who have undertaken research into HSP for several years) or via the British Neurological Surveillance Unit, which regularly communicates with all UK consultant neurologists requesting information on specific patient groups. All individuals gave informed consent to participate in the study, which received multi‐regional ethical committee approval.
Genotyping and linkage analysis
Genomic DNA was extracted from whole blood using standard protocols. Linkage to the SPG7 locus was investigated in the 20 ARHSP families using the polymorphic microsatellite markers D16S413, D16S3023 and D16S303. Amplified polymerase chain reaction (PCR) products were size fractionated by electrophoresis using 8% polyacrylamide gels and visualized by silver staining. Pairwise LOD scores were calculated using MLINK under the assumption of equal allele frequencies and equal male and female recombination rates. Disease inheritance was presumed to be as an autosomal recessive trait with complete penetrance and an allele frequency of 10–4.
Mutation detection
Eighteen pairs of primers were designed, based on chromosome 16 draft sequence data, to amplify the coding region and flanking splice junctions of all 17 exons of the SPG7 gene. PCRs (50 µl) were performed under optimized conditions (Table 1). Single‐stranded conformation polymorphism (SSCP) analysis was performed using 0.6% Mutation Detection Enhancement gels run at 4–10°C for 10–16 h to achieve optimum separation of the strands. Mobility shifts were characterized by direct sequencing of purified PCR products using BIGDYE (Applied Biosystems) and analysed on the ABI 3100 genetic analyser.
Muscle biopsy analysis
Open muscle biopsies were performed under local anaesthetic from vastus lateralis in two patients with SPG7 mutations. Muscle was analysed for standard histochemical reactions including sequential cytochrome c oxidase and succinate dehydrogenase reactions. Muscle samples from both patients, four additional patients with ARHSP in whom linkage to SPG7 had been excluded and 19 age‐matched controls were also prepared and assayed for mitochondrial complex I, II/III, IV and citrate synthase as previously described (Schapira et al., 1990). Activities were corrected for citrate synthase and protein (Biorad), using bovine serum albumin as standard. Respiratory chain complex activity was also measured in mitochondrial fragments isolated from cultured myoblasts by differential centrifugation from both patients and nine age‐matched controls.
Results
Genotyping and linkage analysis
Linkage to the SPG7 locus was excluded in 14 of the 20 ARHSP kindreds (data not shown). The remaining six families were not excluded due to the small pedigree size or uninformative markers. One affected individual from each of these families along with the 29 sporadic spastic paraplegia cases were therefore screened for SPG7 mutations.
Mutation detection
Following initial SSCP analysis and sequencing of all mobility shifts, a total of 12 heterozygous sequence changes were identified in the coding regions of the SPG7 gene. Four were previously reported polymorphisms. Three other conserved sequence changes, 120G→A (G40G), 1816C→T (G605G) and 2283G→A (Q761Q), were also identified in control samples, defining them as novel polymorphisms. The remaining five coding sequence changes were not identified in 200 control chromosomes, supporting these as pathogenic mutations. An additional seven intronic polymorphisms were also identified in patient and control samples. A summary of SPG7 polymorphisms and their relative frequencies in patient and control chromosomes is presented in Table 2.
In total, six SPG7 mutations were identified, five of which were novel (Table 3). Two patients (1 and 2) were compound heterozygotes, each possessing two different mutations. Patient 1 had a family history suggestive of autosomal recessive inheritance, but patient 2 was sporadic. A further sporadic patient (3) initially appeared to carry only a single heterozygous mutation, the 9 bp deletion in exon 11 (1450–1458del9) that had previously been associated with possible autosomal dominant inheritance. Direct sequencing of all the remaining exons in this patient revealed a second sequence change in exon 15, 2026C→T (F676L), which had not produced a visible mobility shift on SSCP under any conditions tested. A new pair of primers containing the sequence change in the first base of the reverse primer were therefore designed, and PCR under optimized conditions successfully amplified the patient DNA but none of 100 control samples, supporting this as the second mutation in this patient.
Clinical characteristics of patients with SPG7 mutations
SPG7 mutations were associated with variably complicated HSP phenotypes in all three patients (Table 4). Age of onset ranged from 11 to 19 years, with slowly progressive deterioration. All demonstrated increased tone in all four limbs, more prominent in the legs, with relatively preserved power. Patients 2 and 3 both had marked cerebellar signs, with limb ataxia, dysarthria and nystagmus. None of the patients had optic atrophy.
MRI brain scans in patients 2 and 3, who had ataxia, showed cerebellar atrophy (Fig. 1). Two small areas of periventricular high T2 signal lesions of uncertain significance were also noted in patient 2. MRI appearances of the spinal cord were normal in all three patients. EMG, nerve conduction studies and visual evoked responses in patients 2 and 3 were also normal.
Both parents of patient 3 had a normal neurological examination as part of this study. Patient 1 was the product of a first cousin marriage, and had four brothers and one sister. By history, one brother may also have been affected. However, no other family members were available for examination, although the parents had previously been examined and were felt to be normal. The parents of patient 2 were also not available (mother died, father declined to be involved), but neither had any neurological or gait abnormalities by history.
Muscle biopsy analysis
Muscle biopsies from patients 2 and 3 both showed changes of denervation with limited reinnervation. In each case, fibre typing showed a marked excess of type 1 fibres with numerous small, atrophic, angulated fibres, predominantly type II. No ragged‐red fibres were seen and, although there were a few scattered cytochrome c oxidase‐negative fibres throughout the biopsies, oxidative stains, including succinate dehydrogenase, were considered within normal limits for the age of the patients.
Analysis of mitochondrial respiratory chain function in muscle from patients 2 and 3 is shown in Fig. 2. Compared with controls, citrate synthase‐corrected activities for complex I and complex II/III fell below the mean control range in both patients, while complex IV activity appeared relatively preserved (Fig. 2A). A similar picture was apparent when the two SPG7 mutation patients were compared with other ARHSP patients in whom linkage to SPG7 had been excluded, with complex I and complex II/III activities falling below mean controls. However, due to the small sample size in this group, confidence intervals for the mean control activities were large and the differences not significant (Table 5). Respiratory chain function assays on mitochondrial fractions isolated from cultured myoblasts in both SPG7 mutation patients also demonstrated a reduction in citrate synthase‐corrected complex I activity outside the standard error for mean control rates, although complex II/III and complex IV activities were within the normal range (Fig. 2B).
Discussion
Since the discovery of SPG7 as the first gene responsible for ARHSP, only four mutations have been reported. Three occurred in consanguineous pedigrees possessing homozygous mutations, while the most recently reported heterozygous 9 bp deletion in exon 11 (1450–1458del9) was found in the father and son from a family in which possible autosomal dominant inheritance was suggested. We have identified six SPG7 mutations, in one case from a family with possible autosomal recessive inheritance, and two apparently sporadic HSP cases. All were compound heterozygotes for two separate mutations. This is unusual for patient 1 who was the product of consanguineous parents. A remarkably similar phenotype with prominent cerebellar ataxia was observed in the two sporadic cases. No mutations were identified in any of the six autosomal recessive HSP patients from families in whom linkage to the SPG7 locus could not be excluded.
Figure 3 shows the distribution of the mutations in SPG7. The two missense mutations in patient 1 occur in highly conserved functional domains. The A10S mutation in exon 1 results in substitution of a hydrophobic with a hydrophilic amino acid residue in the leader peptide of paraplegin. These short amino acid sequences at the N‐terminus of nuclear‐encoded mitochondrial proteins need to form a basic, amphipathic helix in order to allow targeting of the protein to mitochondria (Lithgow, 2000). The introduction of a hydrophilic amino acid residue into the leader peptide is therefore predicted to disrupt the mitochondrial localization of paraplegin. The second mutation, G577S, is situated within the conserved zinc‐binding motif (HESGH), providing further evidence for the functional significance of this domain in paraplegin.
In patient 2, the missense mutation in exon 13 (A572V) results in an amino acid substitution two residues before the zinc‐binding domain. Although both are small non‐polar, hydrophobic amino acids, modelling of the mutant protein with the prediction program GOR4 (http://npsa‐pbil.ibcp.fr/cgi‐bin/secpred_gor4.pl) showed an altered conformation in the region of the zinc‐binding domain from an α‐helix–random coil–α‐helix pattern to an extended strand. This alteration in secondary structure is likely to disrupt the functional activity of this domain. The second mutation in patient 2, a 29 bp deletion in exon 8 (1057–1085del29), is within the AAA cassette involving part of the highly conserved ATP‐binding motif. It causes a frameshift with a premature stop codon at residue 385, predicted to result in a truncated protein.
In patient 3, one mutation was the 9 bp deletion in exon 11 (1450–1458del9), previously reported as a possible autosomal dominant mutation (McDermott et al., 2001). This is within the AAA cassette, resulting in the loss of three amino acids (E,R,R484–486del). SSCP analysis also failed to identify a second mutation in our patient, and it was only by direct sequencing of the remaining exons that a further sequence change, 2026C→T (F676L), was detected. Neurological examination of the parents, both in their 70s and asymptomatic, was entirely normal. Sequencing of the two exons in both parents confirmed that the mother carried the 9 bp deletion in exon 11 and the father the missense mutation in exon 15. In this case, the 1450–1458del9 clearly acts in an autosomal recessive manner with a combination of two heterozygous mutations required to produce the disease phenotype. The F676L substitution did not occur in an obvious functional domain within paraplegin. It results in substitution of leucine for phenylalanine, and this change was not detected in 200 control chromosomes, supporting its role as a pathogenic mutation.
Histological analysis of muscle from two patients with SPG7 mutations failed to demonstrate previously reported morphological changes associated with oxidative phosphorylation impairment, although both were still ambulant and less severely affected than previous cases. The absence of these features on muscle biopsy therefore cannot exclude SPG7 as the cause of the disease. The only consistent finding was of denervation changes with an excess of type I fibres which probably reflects longstanding spasticity.
Mitochondrial respiratory chain activities in muscle from patients 2 and 3, however, showed a reduction in complex I and complex II/III activity compared with controls, when corrected for citrate synthase. Similar results were obtained when compared with other patients with ARHSP in whom SPG7 mutations had been excluded, suggesting that this effect may not simply reflect the effect on muscle of longstanding spasticity and reduced mobility. However, due to the small number of non‐SPG7 ARHSP patients, differences between the two groups did not reach statistical significance.
A previous study of respiratory chain function in muscle from HSP patients included two patients with ARHSP, in whom SPG7 mutations had been excluded (Piemonte et al., 2001). One of these had characteristic histological changes associated with oxidative phosphorylation impairment, and both had isolated complex I deficiencies. A recent study of mitochondrial function in HSP patients in whom both SPG4 and SPG7 mutations had been excluded also demonstrated a reduction in both complex I and complex IV activities (McDermott et al., 2003), suggesting that respiratory chain defects may be common to more than one form of ARHSP.
The pattern of complex I–III deficiency in skeletal muscle is similar to that found in cardiac tissue from patients with Friedreich’s ataxia (Bradley et al., 2000). In Friedreich’s ataxia, deficiency of frataxin is thought to result in abnormal iron–sulphur cluster formation in respiratory chain proteins, leading to oxidative stress and damage. This pattern of complex I–III deficiency is also seen in the SOD2 knockout mouse (Melov et al., 1999). It is therefore possible that the pattern of respiratory chain defect in the muscle from patients with SPG7 mutations may represent excess free radical‐mediated damage. The fact that only a defect in complex I activity was observed in myoblasts may simply represent the accumulation of oxidative damage in a fixed as opposed to dividing tissue.
SPG7 mutations appear to be a relatively rare cause of HSP, identified in only three out of 49 (6%) patients studied. This figure, however, should be regarded as a minimum estimate as SSCP may have failed to detect all mutations. In addition, this study also demonstrates that the SPG7 locus is highly polymorphic. SSCP is not an efficient tool for screening for mutations, and direct sequencing is preferable.
In summary, this study has identified six mutations in three new cases of SPG7 HSP. Biochemical, but not histological changes were found in muscle samples, indicating that standard muscle biopsy staining is not a useful diagnostic test. The presence of ataxia with HSP, even in a sporadic case, should alert the clinician to the possibility of SPG7 mutations as a cause.
The underlying pathogenesis involving mitochondrial dysfunction may make these cases more amenable to therapeutic interventions than other forms of the disease. Further studies are required to determine the precise function of paraplegin in human mitochondria in health and disease in order to facilitate the development of potentially disease‐modifying treatments for this type of HSP.
Acknowledgements
We wish to thank all the patients and their relatives who participated in this study, Dr Angela Brady and the British Neurological Surveillance Unit for referring patients for the study, and Dr Alan Valentine and Dr Mark Cooper for their helpful comments during the preparation of the manuscript. This work was supported by Action Research UK.
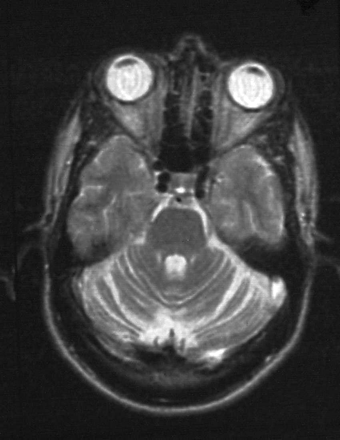
Fig. 1 Axial T2 MRI in patient 3 showing cerebellar atrophy.
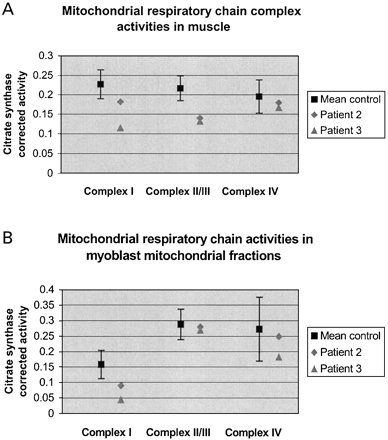
Fig. 2 Respiratory chain complex activity corrected for citrate synthase. Control data are expressed as mean ± 95% confidence interval. Complex IV activity expressed ×10. (A) Homogenized muscle. (B) Myoblast mitochondrial fractions.
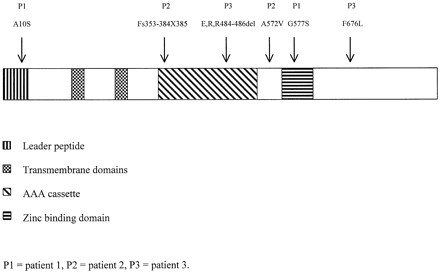
Fig. 3 Schematic representation of the SPG7 gene showing functional domains and sites of mutations.
Frequency of SPG7 polymorphisms in patient and control chromosomes
| Location | Nucleotide change | Predicted protein change | Patient chromosomes (n = 70) | Control chromosomes (n = 100) |
| Exon 1 | 120G→A | G40G | 1 | 1 |
| Intron 4 | IVS4 + 12C→T | – | 36 | 56 |
| Intron 7 | IVS7 + 5G→A | – | 2 | 3 |
| Intron 7 | IVS7 + 17G→C | – | 2 | 2 |
| Intron 7 | IVS7 + 38G→A | – | 3 | 5 |
| Intron 10 | IVS10 + 19G→A | – | 2 | 5 |
| Exon 11 | 1507A→G | T503A | 2 | 6 |
| Exon 11 | 1529C→T | A510V | 1 | 4 |
| Intron 12 | IVS12 + 13C→T | – | 1 | 4 |
| Intron 13 | IVS13 + 45G→C | – | 14 | 31 |
| Exon 14 | 1816C→T | G605G | 0 | 1 |
| Exon 15 | 2063G→A | R688Q | 5 | 24 |
| Exon 17 | 2283G→A | Q761Q | 2 | 2 |
| Exon 17 | 2292C→T | I764I | 2 | 4 |
| Location | Nucleotide change | Predicted protein change | Patient chromosomes (n = 70) | Control chromosomes (n = 100) |
| Exon 1 | 120G→A | G40G | 1 | 1 |
| Intron 4 | IVS4 + 12C→T | – | 36 | 56 |
| Intron 7 | IVS7 + 5G→A | – | 2 | 3 |
| Intron 7 | IVS7 + 17G→C | – | 2 | 2 |
| Intron 7 | IVS7 + 38G→A | – | 3 | 5 |
| Intron 10 | IVS10 + 19G→A | – | 2 | 5 |
| Exon 11 | 1507A→G | T503A | 2 | 6 |
| Exon 11 | 1529C→T | A510V | 1 | 4 |
| Intron 12 | IVS12 + 13C→T | – | 1 | 4 |
| Intron 13 | IVS13 + 45G→C | – | 14 | 31 |
| Exon 14 | 1816C→T | G605G | 0 | 1 |
| Exon 15 | 2063G→A | R688Q | 5 | 24 |
| Exon 17 | 2283G→A | Q761Q | 2 | 2 |
| Exon 17 | 2292C→T | I764I | 2 | 4 |
A = alanine; G = glycine; I = isoleucine; Q = glutamine; R = arginine; T = threonine; V = valine.
Frequency of SPG7 polymorphisms in patient and control chromosomes
| Location | Nucleotide change | Predicted protein change | Patient chromosomes (n = 70) | Control chromosomes (n = 100) |
| Exon 1 | 120G→A | G40G | 1 | 1 |
| Intron 4 | IVS4 + 12C→T | – | 36 | 56 |
| Intron 7 | IVS7 + 5G→A | – | 2 | 3 |
| Intron 7 | IVS7 + 17G→C | – | 2 | 2 |
| Intron 7 | IVS7 + 38G→A | – | 3 | 5 |
| Intron 10 | IVS10 + 19G→A | – | 2 | 5 |
| Exon 11 | 1507A→G | T503A | 2 | 6 |
| Exon 11 | 1529C→T | A510V | 1 | 4 |
| Intron 12 | IVS12 + 13C→T | – | 1 | 4 |
| Intron 13 | IVS13 + 45G→C | – | 14 | 31 |
| Exon 14 | 1816C→T | G605G | 0 | 1 |
| Exon 15 | 2063G→A | R688Q | 5 | 24 |
| Exon 17 | 2283G→A | Q761Q | 2 | 2 |
| Exon 17 | 2292C→T | I764I | 2 | 4 |
| Location | Nucleotide change | Predicted protein change | Patient chromosomes (n = 70) | Control chromosomes (n = 100) |
| Exon 1 | 120G→A | G40G | 1 | 1 |
| Intron 4 | IVS4 + 12C→T | – | 36 | 56 |
| Intron 7 | IVS7 + 5G→A | – | 2 | 3 |
| Intron 7 | IVS7 + 17G→C | – | 2 | 2 |
| Intron 7 | IVS7 + 38G→A | – | 3 | 5 |
| Intron 10 | IVS10 + 19G→A | – | 2 | 5 |
| Exon 11 | 1507A→G | T503A | 2 | 6 |
| Exon 11 | 1529C→T | A510V | 1 | 4 |
| Intron 12 | IVS12 + 13C→T | – | 1 | 4 |
| Intron 13 | IVS13 + 45G→C | – | 14 | 31 |
| Exon 14 | 1816C→T | G605G | 0 | 1 |
| Exon 15 | 2063G→A | R688Q | 5 | 24 |
| Exon 17 | 2283G→A | Q761Q | 2 | 2 |
| Exon 17 | 2292C→T | I764I | 2 | 4 |
A = alanine; G = glycine; I = isoleucine; Q = glutamine; R = arginine; T = threonine; V = valine.
SPG7 mutations detected
| Patient | Location | Nucleotide change | Predicted protein change |
| 1 | Exon 1 | 28G→A | A10S |
| Exon 13 | 1729G→A | G577S | |
| 2 | Exon 8 | 1057–1085del29 | Frameshift 353–384X385 |
| Exon 13 | 1715C→T | A572V | |
| 3 | Exon 11 | 1450–1458del9 | E,R,R484–486del |
| Exon 15 | 2026T→C | F676L |
| Patient | Location | Nucleotide change | Predicted protein change |
| 1 | Exon 1 | 28G→A | A10S |
| Exon 13 | 1729G→A | G577S | |
| 2 | Exon 8 | 1057–1085del29 | Frameshift 353–384X385 |
| Exon 13 | 1715C→T | A572V | |
| 3 | Exon 11 | 1450–1458del9 | E,R,R484–486del |
| Exon 15 | 2026T→C | F676L |
A = alanine; E = glutamic acid; F = phenylalanine; G = glycine; L = leucine; R = arginine; S = serine; V = valine.
SPG7 mutations detected
| Patient | Location | Nucleotide change | Predicted protein change |
| 1 | Exon 1 | 28G→A | A10S |
| Exon 13 | 1729G→A | G577S | |
| 2 | Exon 8 | 1057–1085del29 | Frameshift 353–384X385 |
| Exon 13 | 1715C→T | A572V | |
| 3 | Exon 11 | 1450–1458del9 | E,R,R484–486del |
| Exon 15 | 2026T→C | F676L |
| Patient | Location | Nucleotide change | Predicted protein change |
| 1 | Exon 1 | 28G→A | A10S |
| Exon 13 | 1729G→A | G577S | |
| 2 | Exon 8 | 1057–1085del29 | Frameshift 353–384X385 |
| Exon 13 | 1715C→T | A572V | |
| 3 | Exon 11 | 1450–1458del9 | E,R,R484–486del |
| Exon 15 | 2026T→C | F676L |
A = alanine; E = glutamic acid; F = phenylalanine; G = glycine; L = leucine; R = arginine; S = serine; V = valine.
Clinical characteristics of patients with SPG7 mutations
| Patient | 1 | 2 | 3 |
| Age (years) | 21 | 51 | 42 |
| Sex | M | F | M |
| Age at onset (years) | 11 | 14 | 19 |
| Dysarthria | – | + | + |
| Nystagmus | – | + | + |
| Bladder dysfunction | – | – | + |
| Upper limb | |||
| Spasticity | + | + | + |
| Weakness | – | – | – |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | – | – |
| Lower limb | |||
| Pes cavus | + | + | – |
| Spasticity | + | + | + |
| Weakness | – | + | + |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | + | – |
| Plantar reflexes | ↑↑ | ↑↑ | ↑↑ |
| Gait | Spastic | Spastic–ataxic | Spastic–ataxic |
| Functional status | Independent | Requires stick | Independent |
| EMG/NCS | Not done | Normal | Normal |
| MRI brain | Normal | Periventricular T2 hyperintensity. Cerebellar atrophy | Cerebellar atrophy |
| MRI spinal cord | Normal | Normal | Normal |
| Patient | 1 | 2 | 3 |
| Age (years) | 21 | 51 | 42 |
| Sex | M | F | M |
| Age at onset (years) | 11 | 14 | 19 |
| Dysarthria | – | + | + |
| Nystagmus | – | + | + |
| Bladder dysfunction | – | – | + |
| Upper limb | |||
| Spasticity | + | + | + |
| Weakness | – | – | – |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | – | – |
| Lower limb | |||
| Pes cavus | + | + | – |
| Spasticity | + | + | + |
| Weakness | – | + | + |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | + | – |
| Plantar reflexes | ↑↑ | ↑↑ | ↑↑ |
| Gait | Spastic | Spastic–ataxic | Spastic–ataxic |
| Functional status | Independent | Requires stick | Independent |
| EMG/NCS | Not done | Normal | Normal |
| MRI brain | Normal | Periventricular T2 hyperintensity. Cerebellar atrophy | Cerebellar atrophy |
| MRI spinal cord | Normal | Normal | Normal |
M = male; F = female; + = present; – = absent; ↑↑= bilaterally extensor; NCS = nerve conduction studies.
Clinical characteristics of patients with SPG7 mutations
| Patient | 1 | 2 | 3 |
| Age (years) | 21 | 51 | 42 |
| Sex | M | F | M |
| Age at onset (years) | 11 | 14 | 19 |
| Dysarthria | – | + | + |
| Nystagmus | – | + | + |
| Bladder dysfunction | – | – | + |
| Upper limb | |||
| Spasticity | + | + | + |
| Weakness | – | – | – |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | – | – |
| Lower limb | |||
| Pes cavus | + | + | – |
| Spasticity | + | + | + |
| Weakness | – | + | + |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | + | – |
| Plantar reflexes | ↑↑ | ↑↑ | ↑↑ |
| Gait | Spastic | Spastic–ataxic | Spastic–ataxic |
| Functional status | Independent | Requires stick | Independent |
| EMG/NCS | Not done | Normal | Normal |
| MRI brain | Normal | Periventricular T2 hyperintensity. Cerebellar atrophy | Cerebellar atrophy |
| MRI spinal cord | Normal | Normal | Normal |
| Patient | 1 | 2 | 3 |
| Age (years) | 21 | 51 | 42 |
| Sex | M | F | M |
| Age at onset (years) | 11 | 14 | 19 |
| Dysarthria | – | + | + |
| Nystagmus | – | + | + |
| Bladder dysfunction | – | – | + |
| Upper limb | |||
| Spasticity | + | + | + |
| Weakness | – | – | – |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | – | – |
| Lower limb | |||
| Pes cavus | + | + | – |
| Spasticity | + | + | + |
| Weakness | – | + | + |
| Ataxia | – | + | + |
| Hyperreflexia | + | + | + |
| Sensory impairment | – | + | – |
| Plantar reflexes | ↑↑ | ↑↑ | ↑↑ |
| Gait | Spastic | Spastic–ataxic | Spastic–ataxic |
| Functional status | Independent | Requires stick | Independent |
| EMG/NCS | Not done | Normal | Normal |
| MRI brain | Normal | Periventricular T2 hyperintensity. Cerebellar atrophy | Cerebellar atrophy |
| MRI spinal cord | Normal | Normal | Normal |
M = male; F = female; + = present; – = absent; ↑↑= bilaterally extensor; NCS = nerve conduction studies.
Mitochondrial respiratory chain complex activities corrected for citrate synthase in SPG7 mutation patients, non‐SPG7 ARHSP patients and controls
| Complex I | Complex II/III | Complex IV | |
| Muscle | |||
| Patient 2 | 0.182 | 0.132 | 0.018 |
| Patient 3 | 0.115 | 0.14 | 0.017 |
| Controls | 0.227 ± 0.037 | 0.217 ± 0.032 | 0.0196 ± 0.0043 |
| Non‐SPG7 ARHSP | 0.185 ± 0.085 | 0.211 ± 0.116 | 0.171 ± 0.012 |
| Myoblast | |||
| Patient 2 | 0.09 | 0.27 | 0.025 |
| Patient 3 | 0.044 | 0.28 | 0.018 |
| Controls | 0.158 ± 0.046 | 0.288 ± 0.046 | 0.027 ± 0.011 |
| Complex I | Complex II/III | Complex IV | |
| Muscle | |||
| Patient 2 | 0.182 | 0.132 | 0.018 |
| Patient 3 | 0.115 | 0.14 | 0.017 |
| Controls | 0.227 ± 0.037 | 0.217 ± 0.032 | 0.0196 ± 0.0043 |
| Non‐SPG7 ARHSP | 0.185 ± 0.085 | 0.211 ± 0.116 | 0.171 ± 0.012 |
| Myoblast | |||
| Patient 2 | 0.09 | 0.27 | 0.025 |
| Patient 3 | 0.044 | 0.28 | 0.018 |
| Controls | 0.158 ± 0.046 | 0.288 ± 0.046 | 0.027 ± 0.011 |
±95% confidence interval.
Mitochondrial respiratory chain complex activities corrected for citrate synthase in SPG7 mutation patients, non‐SPG7 ARHSP patients and controls
| Complex I | Complex II/III | Complex IV | |
| Muscle | |||
| Patient 2 | 0.182 | 0.132 | 0.018 |
| Patient 3 | 0.115 | 0.14 | 0.017 |
| Controls | 0.227 ± 0.037 | 0.217 ± 0.032 | 0.0196 ± 0.0043 |
| Non‐SPG7 ARHSP | 0.185 ± 0.085 | 0.211 ± 0.116 | 0.171 ± 0.012 |
| Myoblast | |||
| Patient 2 | 0.09 | 0.27 | 0.025 |
| Patient 3 | 0.044 | 0.28 | 0.018 |
| Controls | 0.158 ± 0.046 | 0.288 ± 0.046 | 0.027 ± 0.011 |
| Complex I | Complex II/III | Complex IV | |
| Muscle | |||
| Patient 2 | 0.182 | 0.132 | 0.018 |
| Patient 3 | 0.115 | 0.14 | 0.017 |
| Controls | 0.227 ± 0.037 | 0.217 ± 0.032 | 0.0196 ± 0.0043 |
| Non‐SPG7 ARHSP | 0.185 ± 0.085 | 0.211 ± 0.116 | 0.171 ± 0.012 |
| Myoblast | |||
| Patient 2 | 0.09 | 0.27 | 0.025 |
| Patient 3 | 0.044 | 0.28 | 0.018 |
| Controls | 0.158 ± 0.046 | 0.288 ± 0.046 | 0.027 ± 0.011 |
±95% confidence interval.
Primers and conditions for SPG7 PCRs
| Exon | Forward primer | Reverse primer | Annealing temperatue (°C) | mM MgCl2 | PCR product size (bp) |
| 1 | 5′ ATCACGCAGGCGCGGCTTTCAG 3′ | 5′ CTGGGCCTTACAGAGCAGA 3′ | 60 | 1.5 | 270 |
| 2 | 5′ AGTCTGCATTGCTTTGGTACT 3′ | 5′ TAGCTGAGGCGATAAGTGTG 3′ | 57 | 1.5 | 228 |
| 3 | 5′ GGAGTACACTGTTGTCCTGT 3′ | 5′ ACAGAAATGTAAAGACATCCAG 3′ | 55 | 1.5 | 226 |
| 4A | 5′ AAGCTCTGGATGTCGCCCGT 3′ | 5′ AGGAAATGCTGCCTCCGCTG 3′ | 57 | 1.5 | 193 |
| 4B | 5′ GCGGTTGTCATGAGCCTCCT 3′ | 5′ CTCACTCTCACAGGCTGCCA 3′ | 57 | 2.0 | 241 |
| 5 | 5′ GACTGTAGGGTTGCTCGTCT 3′ | 5′ CAGATTACAAAGCCAAGTTAGG 3′ | 55 | 1.5 | 260 |
| 6 | 5′ TTGGAAGCCTGCGTCTGTCA 3′ | 5′ GTATTCAGCAAACACAAACCAG 3′ | 57 | 1.5 | 225 |
| 7 | 5′ CTGGCATCGTGCTGCTGATT 3′ | 5′ CCCTTCTGGGAGAGGAGGA 3′ | 57 | 1.5 | 257 |
| 8 | 5′ AGTGTTGCATTGTCTGCTGC 3′ | 5′ ATGTGTGAAAGGAGCCAGGT 3′ | 57 | 2.0 | 252 |
| 9A | 5′ CCTTGGTGTAGAACTTTGTCT 3′ | 5′ TGTTGGAGAAGCCGGACATG 3′ | 55 | 1.5 | 221 |
| 9B | 5′ GCATCGTCTACATCGATGAG 3′ | 5′ CCTGTTCTGAAAGACATCGG 3′ | 55 | 1.5 | 187 |
| 10 | 5′ TCCCTCCTGTGTCCTGAAGG3′ | 5′ CCAGACCACTCAGAGCGAGT 3′ | 57 | 1.5 | 283 |
| 11 | 5′ ACCTGTGGCAGTAACTAGGT 3′ | 5′ GCCTTGATGCTGTTTGCGCA 3′ | 57 | 1.5 | 211 |
| 12 | 5′ CTCTTAAGCCCTGATAGCAG 3′ | 5′ TCACCTCTCAATACCTGCCT 3′ | 55 | 2.0 | 252 |
| 13 | 5′ GTCTCGAACTCCTGTCCTCA 3′ | 5′ AGTCAGCTACAGACACAGGC 3′ | 60 | 1.5 | 300 |
| 14 | 5′ ACGGAGACCTCTTAGTCCCA 3′ | 5′ CATGGCATGCACTGGAACAG 3′ | 55 | 2.0 | 321 |
| 15 | 5′ ACTGCTCTGCGCCTGCAGT 3′ | 5′ CCTTGTGTGGTAGACCCA 3′ | 57 | 1.5 | 294 |
| 16 | 5′ TCTGTGCTTTGGTGCTGGAG 3′ | 5′ ACCGTGGGTGCTGTGTGGA 3′ | 57 | 1.5 | 206 |
| 17 | 5′ ACATGCATATGCCTGTTCTTT 3′ | 5′ CTCAGCTGAAAAGCAACTCAG 3′ | 55 | 1.5 | 312 |
| Exon | Forward primer | Reverse primer | Annealing temperatue (°C) | mM MgCl2 | PCR product size (bp) |
| 1 | 5′ ATCACGCAGGCGCGGCTTTCAG 3′ | 5′ CTGGGCCTTACAGAGCAGA 3′ | 60 | 1.5 | 270 |
| 2 | 5′ AGTCTGCATTGCTTTGGTACT 3′ | 5′ TAGCTGAGGCGATAAGTGTG 3′ | 57 | 1.5 | 228 |
| 3 | 5′ GGAGTACACTGTTGTCCTGT 3′ | 5′ ACAGAAATGTAAAGACATCCAG 3′ | 55 | 1.5 | 226 |
| 4A | 5′ AAGCTCTGGATGTCGCCCGT 3′ | 5′ AGGAAATGCTGCCTCCGCTG 3′ | 57 | 1.5 | 193 |
| 4B | 5′ GCGGTTGTCATGAGCCTCCT 3′ | 5′ CTCACTCTCACAGGCTGCCA 3′ | 57 | 2.0 | 241 |
| 5 | 5′ GACTGTAGGGTTGCTCGTCT 3′ | 5′ CAGATTACAAAGCCAAGTTAGG 3′ | 55 | 1.5 | 260 |
| 6 | 5′ TTGGAAGCCTGCGTCTGTCA 3′ | 5′ GTATTCAGCAAACACAAACCAG 3′ | 57 | 1.5 | 225 |
| 7 | 5′ CTGGCATCGTGCTGCTGATT 3′ | 5′ CCCTTCTGGGAGAGGAGGA 3′ | 57 | 1.5 | 257 |
| 8 | 5′ AGTGTTGCATTGTCTGCTGC 3′ | 5′ ATGTGTGAAAGGAGCCAGGT 3′ | 57 | 2.0 | 252 |
| 9A | 5′ CCTTGGTGTAGAACTTTGTCT 3′ | 5′ TGTTGGAGAAGCCGGACATG 3′ | 55 | 1.5 | 221 |
| 9B | 5′ GCATCGTCTACATCGATGAG 3′ | 5′ CCTGTTCTGAAAGACATCGG 3′ | 55 | 1.5 | 187 |
| 10 | 5′ TCCCTCCTGTGTCCTGAAGG3′ | 5′ CCAGACCACTCAGAGCGAGT 3′ | 57 | 1.5 | 283 |
| 11 | 5′ ACCTGTGGCAGTAACTAGGT 3′ | 5′ GCCTTGATGCTGTTTGCGCA 3′ | 57 | 1.5 | 211 |
| 12 | 5′ CTCTTAAGCCCTGATAGCAG 3′ | 5′ TCACCTCTCAATACCTGCCT 3′ | 55 | 2.0 | 252 |
| 13 | 5′ GTCTCGAACTCCTGTCCTCA 3′ | 5′ AGTCAGCTACAGACACAGGC 3′ | 60 | 1.5 | 300 |
| 14 | 5′ ACGGAGACCTCTTAGTCCCA 3′ | 5′ CATGGCATGCACTGGAACAG 3′ | 55 | 2.0 | 321 |
| 15 | 5′ ACTGCTCTGCGCCTGCAGT 3′ | 5′ CCTTGTGTGGTAGACCCA 3′ | 57 | 1.5 | 294 |
| 16 | 5′ TCTGTGCTTTGGTGCTGGAG 3′ | 5′ ACCGTGGGTGCTGTGTGGA 3′ | 57 | 1.5 | 206 |
| 17 | 5′ ACATGCATATGCCTGTTCTTT 3′ | 5′ CTCAGCTGAAAAGCAACTCAG 3′ | 55 | 1.5 | 312 |
Primers and conditions for SPG7 PCRs
| Exon | Forward primer | Reverse primer | Annealing temperatue (°C) | mM MgCl2 | PCR product size (bp) |
| 1 | 5′ ATCACGCAGGCGCGGCTTTCAG 3′ | 5′ CTGGGCCTTACAGAGCAGA 3′ | 60 | 1.5 | 270 |
| 2 | 5′ AGTCTGCATTGCTTTGGTACT 3′ | 5′ TAGCTGAGGCGATAAGTGTG 3′ | 57 | 1.5 | 228 |
| 3 | 5′ GGAGTACACTGTTGTCCTGT 3′ | 5′ ACAGAAATGTAAAGACATCCAG 3′ | 55 | 1.5 | 226 |
| 4A | 5′ AAGCTCTGGATGTCGCCCGT 3′ | 5′ AGGAAATGCTGCCTCCGCTG 3′ | 57 | 1.5 | 193 |
| 4B | 5′ GCGGTTGTCATGAGCCTCCT 3′ | 5′ CTCACTCTCACAGGCTGCCA 3′ | 57 | 2.0 | 241 |
| 5 | 5′ GACTGTAGGGTTGCTCGTCT 3′ | 5′ CAGATTACAAAGCCAAGTTAGG 3′ | 55 | 1.5 | 260 |
| 6 | 5′ TTGGAAGCCTGCGTCTGTCA 3′ | 5′ GTATTCAGCAAACACAAACCAG 3′ | 57 | 1.5 | 225 |
| 7 | 5′ CTGGCATCGTGCTGCTGATT 3′ | 5′ CCCTTCTGGGAGAGGAGGA 3′ | 57 | 1.5 | 257 |
| 8 | 5′ AGTGTTGCATTGTCTGCTGC 3′ | 5′ ATGTGTGAAAGGAGCCAGGT 3′ | 57 | 2.0 | 252 |
| 9A | 5′ CCTTGGTGTAGAACTTTGTCT 3′ | 5′ TGTTGGAGAAGCCGGACATG 3′ | 55 | 1.5 | 221 |
| 9B | 5′ GCATCGTCTACATCGATGAG 3′ | 5′ CCTGTTCTGAAAGACATCGG 3′ | 55 | 1.5 | 187 |
| 10 | 5′ TCCCTCCTGTGTCCTGAAGG3′ | 5′ CCAGACCACTCAGAGCGAGT 3′ | 57 | 1.5 | 283 |
| 11 | 5′ ACCTGTGGCAGTAACTAGGT 3′ | 5′ GCCTTGATGCTGTTTGCGCA 3′ | 57 | 1.5 | 211 |
| 12 | 5′ CTCTTAAGCCCTGATAGCAG 3′ | 5′ TCACCTCTCAATACCTGCCT 3′ | 55 | 2.0 | 252 |
| 13 | 5′ GTCTCGAACTCCTGTCCTCA 3′ | 5′ AGTCAGCTACAGACACAGGC 3′ | 60 | 1.5 | 300 |
| 14 | 5′ ACGGAGACCTCTTAGTCCCA 3′ | 5′ CATGGCATGCACTGGAACAG 3′ | 55 | 2.0 | 321 |
| 15 | 5′ ACTGCTCTGCGCCTGCAGT 3′ | 5′ CCTTGTGTGGTAGACCCA 3′ | 57 | 1.5 | 294 |
| 16 | 5′ TCTGTGCTTTGGTGCTGGAG 3′ | 5′ ACCGTGGGTGCTGTGTGGA 3′ | 57 | 1.5 | 206 |
| 17 | 5′ ACATGCATATGCCTGTTCTTT 3′ | 5′ CTCAGCTGAAAAGCAACTCAG 3′ | 55 | 1.5 | 312 |
| Exon | Forward primer | Reverse primer | Annealing temperatue (°C) | mM MgCl2 | PCR product size (bp) |
| 1 | 5′ ATCACGCAGGCGCGGCTTTCAG 3′ | 5′ CTGGGCCTTACAGAGCAGA 3′ | 60 | 1.5 | 270 |
| 2 | 5′ AGTCTGCATTGCTTTGGTACT 3′ | 5′ TAGCTGAGGCGATAAGTGTG 3′ | 57 | 1.5 | 228 |
| 3 | 5′ GGAGTACACTGTTGTCCTGT 3′ | 5′ ACAGAAATGTAAAGACATCCAG 3′ | 55 | 1.5 | 226 |
| 4A | 5′ AAGCTCTGGATGTCGCCCGT 3′ | 5′ AGGAAATGCTGCCTCCGCTG 3′ | 57 | 1.5 | 193 |
| 4B | 5′ GCGGTTGTCATGAGCCTCCT 3′ | 5′ CTCACTCTCACAGGCTGCCA 3′ | 57 | 2.0 | 241 |
| 5 | 5′ GACTGTAGGGTTGCTCGTCT 3′ | 5′ CAGATTACAAAGCCAAGTTAGG 3′ | 55 | 1.5 | 260 |
| 6 | 5′ TTGGAAGCCTGCGTCTGTCA 3′ | 5′ GTATTCAGCAAACACAAACCAG 3′ | 57 | 1.5 | 225 |
| 7 | 5′ CTGGCATCGTGCTGCTGATT 3′ | 5′ CCCTTCTGGGAGAGGAGGA 3′ | 57 | 1.5 | 257 |
| 8 | 5′ AGTGTTGCATTGTCTGCTGC 3′ | 5′ ATGTGTGAAAGGAGCCAGGT 3′ | 57 | 2.0 | 252 |
| 9A | 5′ CCTTGGTGTAGAACTTTGTCT 3′ | 5′ TGTTGGAGAAGCCGGACATG 3′ | 55 | 1.5 | 221 |
| 9B | 5′ GCATCGTCTACATCGATGAG 3′ | 5′ CCTGTTCTGAAAGACATCGG 3′ | 55 | 1.5 | 187 |
| 10 | 5′ TCCCTCCTGTGTCCTGAAGG3′ | 5′ CCAGACCACTCAGAGCGAGT 3′ | 57 | 1.5 | 283 |
| 11 | 5′ ACCTGTGGCAGTAACTAGGT 3′ | 5′ GCCTTGATGCTGTTTGCGCA 3′ | 57 | 1.5 | 211 |
| 12 | 5′ CTCTTAAGCCCTGATAGCAG 3′ | 5′ TCACCTCTCAATACCTGCCT 3′ | 55 | 2.0 | 252 |
| 13 | 5′ GTCTCGAACTCCTGTCCTCA 3′ | 5′ AGTCAGCTACAGACACAGGC 3′ | 60 | 1.5 | 300 |
| 14 | 5′ ACGGAGACCTCTTAGTCCCA 3′ | 5′ CATGGCATGCACTGGAACAG 3′ | 55 | 2.0 | 321 |
| 15 | 5′ ACTGCTCTGCGCCTGCAGT 3′ | 5′ CCTTGTGTGGTAGACCCA 3′ | 57 | 1.5 | 294 |
| 16 | 5′ TCTGTGCTTTGGTGCTGGAG 3′ | 5′ ACCGTGGGTGCTGTGTGGA 3′ | 57 | 1.5 | 206 |
| 17 | 5′ ACATGCATATGCCTGTTCTTT 3′ | 5′ CTCAGCTGAAAAGCAACTCAG 3′ | 55 | 1.5 | 312 |
References
Banfi S, Bassi MT, Andolfi G, Marchitiello A, Zanotta S, Ballabio A, et al. Identification and characterization of AFG3L2, a novel paraplegin‐related gene.
Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira, AH. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia.
Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear‐encoded mitochondrial metalloprotease.
Coppola M, Pizzigoni A, Banfi S, Bassi MT, Casari G, Incerti B. Identification and characterization of YME1L1, a novel paraplegin‐related gene.
De Michele G, De Fusco M, Cavalcanti F, Filla A, Marconi R, Volpe G, et al. A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3.
Hansen JJ, Durr A, Cournu‐Rebeix I, Georgopoulos C, Ang D, Nielsen MN, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60.
Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia.
Kremmidiotis G, Gardner AE, Settasatian C, Savoia A, Sutherland GR, Callen DF. Molecular and functional analyses of the human and mouse genes encoding AFG3L1, a mitochondrial metalloprotease homologous to the human spastic paraplegia protein.
McDermott CJ, Dayaratne RK, Tomkins J, Lusher ME, Lindsey JC, Johnson MA, et al. Paraplegin gene analysis in hereditary spastic paraparesis (HSP) pedigrees in northeast England.
McDermott CJ, Taylor RW, Hayes C, Johnson M, Bushby, KM, Turnbull DM, et al. Investigation of mitochondrial function in hereditary spastic paraparesis.
Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice.
Patel S, Latterich M. The AAA team: related ATPases with diverse functions.
Patel H, Cross H, Proukakis C, Hershberger R, Bork P, Ciccarelli FD, et al. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia.
Paul MF, Tzagoloff A. Mutations in RCA1 and AFG3 inhibit F1‐ATPase assembly in Saccharomyces cerevisiae.
Pearce DA. Hereditary spastic paraplegia: mitochondrial metalloproteases of yeast.
Piemonte F, Casali C, Carrozzo R, Schagger H, Patrono C, Tessa A, et al. Respiratory chain defects in hereditary spastic paraplegias.
Polo JM, Calleja J, Combarros O, Berciano J. Hereditary ataxias and paraplegias in Cantabria, Spain. An epidemiological and clinical study.
Polo JM, Calleja J, Combarros O, Berciano J. Hereditary ‘pure’ spastic paraplegia: a study of nine families.
Rainier S, Chai J‐H, Tokarz D, Nicholls RD, Fink JK. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6).
Reid E, Kloos M, Ashley‐Koch A, Hughes L, Bevan S, Svenson IK, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10).
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease.
Settasatian C, Whitmore SA, Crawford J, Bilton RL, Cleton‐Jansen AM, Sutherland GR, et al. Genomic structure and expression analysis of the spastic paraplegia gene, SPG7.
Sridharan R, Radhakrishnan K, Ashok PP, Mousa ME. Prevalence and pattern of spinocerebellar degenerations in northeastern Libya.


{kind=link}
{kind=link}
{kind=link}