




Abstract
Traumatic brain injury is the most common cause of death and disability in young people and survivors often suffer from chronic cognitive deficits. From animal, post-mortem and cognitive studies, there is now increased evidence that abnormalities in the cholinergic system may be underlying some of these deficits. This study investigated this hypothesis in a group of survivors of moderate–severe head injury (n = 31). Patients completed a comprehensive neuropsychological assessment and an MRI scan. Compared with a group of controls (matched on age, sex and premorbid intelligence quotient), the patients showed deficits in sustained attention, paired associate learning and reaction time, but comparative preservation of spatial working memory. Voxel-based morphometry revealed reduced grey matter density in the head injured group in the basal forebrain, the hippocampal formation and regions of the neocortex. These cognitive and structural results are consistent with cholinergic dysfunction. These preliminary findings suggest that cholinergic enhancers may be an effective treatment of cognitive deficits post head injury.
Introduction
Traumatic brain injury is the most common cause of death and disability in young people (Ghajar, 2000) and is sustained by ∼0.5–1 million individuals each year (Field, 1975; Jennett, 1996). Advances in critical care, imaging and trauma system organizations have led to a decrease in mortality from head injury over the last 25 years (Ghajar, 2000). However, survivors of head injury often suffer from chronic cognitive deficits. The neurochemical substrates underlying these deficits are not well understood.
There is now increasing evidence suggesting that abnormalities in the cholinergic system may contribute to these cognitive sequelae (see Arciniegas, 2003). The cholinergic system is made up of a series of nuclei, predominantly located in the basal forebrain, which maintain discrete terminal fields and projections (see Fig. 1; Hanaway et al., 1998; Selden et al., 1998; Perry et al., 1999). There are three main strands of evidence linking acetylcholine (ACh) to cognitive deficits post head injury.
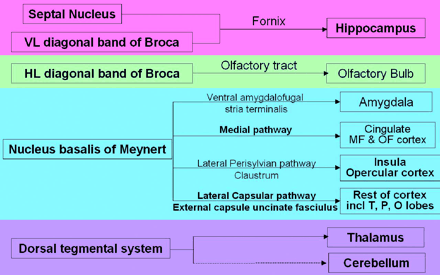
Major ACh pathways (based on Hanaway et al., 1998; Selden et al., 1998; Perry et al., 1999). Bold, script indicates abnormality detected using VBM in this study. MF = Medial frontal; OF = Orbito frontal; O = Occipital; P = Parietal; T = Temporal.
The neuropsychological deficits associated with head injury are in those functions known to be modulated by cholinergic mechanisms. Pathological studies post head injury in humans and animal models suggest chronic cholinergic abnormalities in the presence of relative normality of other neurotransmitters and the results of pharmacological interventions post head injury are consistent with dysfunction of the cholinergic pathways.
Neuropsychological deficits following head injury
The incidence of cognitive deficits post head injury is not consistently documented, with many outcome studies relying on insensitive gross measures such as the Glasgow Outcome Scale (e.g. Signorini et al., 1999; Ono et al., 2001). Further, considerable variation in the severity of head injury sustained by study samples makes cross study comparisons complex. Despite these caveats, impairments in attention and memory appear to be relatively consistent findings (Arciniegas et al., 1999). Deficits in memory and sustained, selective and divided attention and delayed reaction times have been reported following severe, moderate and mild head injury (Arcia and Gualtieri, 1994; Whyte et al., 1995; Robertson et al., 1997; Hellawell et al., 1999; Chan, 2000; Polo et al., 2002). The significance of these deficits is underlined by their impact on the patients' everyday life (see Robertson et al., 1997; Chan, 2000; Fortin et al., 2003).
Acetylcholine has been associated with attentional performance in a number of different studies. In humans, administration of ACh modulators (including nicotine and scopolamine) modulates sustained attention performance (Rezvani and Levin, 2001; see Sahakian, 1988 for review). Microdialysis studies in rats have revealed that ACh is released during sustained attention, with increased releases associated with increasing difficulty (Himmelheber et al., 2000; Dalley et al., 2001). Acetylcholine inputs and projections are critical for the neuronal changes associated with sustained attention in the medial prefrontal cortex (Gill et al., 2000). Further, lesions to the basal forebrain cholinergic system in rats and monkeys impair attentional performance (Muir et al., 1994; Voytko et al., 1994). A similar deficit was also induced by infusing the basal forebrain cholinergic system with muscimol (a GABA agonist which inhibits ACh cells) (Muir et al., 1992).
Acetylcholine has also been implicated in memory performance. In humans, scopolamine administration disrupts acquisition and recall of information, especially visuospatial information (Crow and Grove-White, 1973; Kopelman, 1986). In contrast, nicotine improves memory performance (Rusted et al., 1995). Furthermore, cholinesterase inhibitors have been shown to be effective in the treatment of the cognitive symptoms of attention and mnemonic impairments in patients with Alzheimer's disease (Vesey et al., 2002). Pharmacological manipulation of the ACh system also modulates memory performance in animals (e.g. Ridley et al., 1984; Levin et al., 2003; Rogers and Kesner, 2003). Levels of ACh release are closely associated with levels of memory performance (Ragozzino et al., 1996) and the process of consolidation appears to be associated with decreases in ACh levels (Hasselmo, 1999). Finally, memory deficits following basal forebrain damage have been reported in both humans (Damasio et al., 1985; Abe et al., 1998) and animals (Voytko, 1996; Fine et al., 1997; Ridley et al., 1988, 1989, 1999; Barefoot et al., 2000). It should be emphasized that these lesions are likely to include non-cholinergic neurons, meaning that the deficits observed may not be solely attributable to ACh. Nevertheless, the importance of the cholinergic system is emphasized by the finding that the memory impairment in lesioned animals is reversed following administration of an ACh agonist (Ridley et al., 1988, 1989; Barefoot et al., 2000).
Neuropathology studies post head injury
Pathological studies of ACh function in humans post head injury are currently limited to studies of post-mortem brain tissue. Two studies have reported widespread presynaptic cholinergic dysfunction in the neocortex (Dewar and Graham, 1996; Murdoch et al., 1998). MRI has been used in survivors of head injury to examine the structural pattern of pathology. However, many of these studies have relied on insensitive measures of pathology or have focussed on isolated brain regions (e.g. van der Naalt et al., 1999), making interpretation of the data difficult. Murdoch et al. (2002) reported damage to the nucleus basalis of Meynert following fatal head injury. The possibility that a similar pattern of damage (although potentially less severe) is seen in survivors of head injury is supported by the studies of animal models of head injury.
Reductions in choline acetyltransferase (CHAT) positive neurons in the basal forebrain have been reported in animals post fluid percussion injury (Leonard et al., 1994; Dixon et al., 1997a; Pike and Hamm, 1997; Sinson et al., 1997), and are accompanied by reduced CHAT activity (Gorman et al., 1996; Schmidt et al., 1999). Other abnormalities associated with head injury in animals include reduced high-affinity choline uptake (Dixon et al., 1994a), decreased levels of ACh (as measured by microdialysis) (Dixon et al., 1995, 1997b) and abnormalities in muscarinic receptors (Delahunty, 1992; Delahunty et al., 1994; DeAngelis et al., 1994; Jiang et al., 1994; Lyeth et al., 1994). The anatomical regions most frequently damaged in animal models of head injury are the hippocampal formation (Leonard et al., 1997; Sihver et al., 2001; Chen et al., 2003) and the thalamus (Chen et al., 2003). These areas are heavily innervated by ACh projections in the normal brain (see Bentivoglio and Steriade, 1990).
Pharmacological manipulations post head injury
Although there are currently no approved pharmacological strategies for treatment of cognitive deficits post head injury (Narayan et al., 2002), there are a few reports of improved cognition following manipulation of the ACh pathways. In single cases, administration of physostigmine has been reported to improve memory (Goldberg et al., 1982), attention (Levin et al., 1986) and arousal (Weinberg et al., 1987) in survivors of head injury. Such amelioriation of cognitive sequelae is consistent with the hypothesis that ACh deficits underlie these difficulties. Further evidence of disruption of the ACh system in survivors of head injury comes from reports of low tolerance of ACh antagonist (Sandel et al., 1993; Stanislav, 1997; Müller et al., 1999). Such a pattern (which is also seen in animal models of head injury (e.g. Dixon et al., 1994b; Voytko et al., 1994; Fine et al., 1997)) is suggestive of a decreased reserve in cholinergic function in these individuals (Arciniegas et al., 1999). Indeed, head injury is a risk factor for Alzheimer's disease, a disease with a relatively selective dysfunction of the cholinergic system at least in the early stages of the disease (Heyman et al., 1984; Mortimer et al., 1985; French et al., 1985; Graves et al., 1990; Rasmusson et al., 1995; Nemetz et al., 1999; Lye and Shores, 2000; Fleminger et al., 2003; but see Shalat et al., 1987; Broe et al., 1990). The role of apolipoprotein E (ApoE) genotype in this increased risk remains unclear (e.g. Millar et al., 2003).
In light of this evidence for a role for ACh in the cognitive sequelae of head injury, this study sought to obtain a comprehensive neuropsychological profile of survivors of head injury, using assessments known to be sensitive to cholinergic dysfunction. In addition, MRI scans were obtained and analysed using voxel-based morphometry (VBM) in order to search throughout the brain for structural changes consistent with abnormalities of the cholinergic system in survivors of head injury.
Material and methods
This study had ethical approval from the Cambridge Local Research Ethics Committee (LREC 97/270) and consent was obtained according to the Declaration of Helsinki.
Patients and control participants
Thirty-one patients were recruited from a group of patients suffering from traumatic head injury who required admission to the Neurosciences Critical Care Unit, Addenbrooke's Hospital, Cambridge. Individuals suffering from neurodegenerative diseases were excluded. All patients were at least 6 months post injury. Thirty-two control volunteers were recruited. Individuals (both patients and control volunteers) with a prior history of contact with neurological or psychiatric services were excluded. Individuals (in either group) with a history of alcoholism (including pre- and post-injury) were excluded from the study (to rule out the possibility that findings were attributable to neural changes associated with alcoholism – see Weiss and Porrino, 2002). Premorbid full-scale intelligence quotient (IQ) was measured using the National Adult Reading Test (NART) (Nelson, 1982) and participants completed the Mini Mental State Examination (MMSE) (Folstein et al., 1975) and the Beck Depression Inventory (BDI) (Beck, 1970).
The sample was compared with previously published cohort Neuroscience Critical Care Unit data to determine how representative of the entire intensive care treated head injury population it was (Patel et al., 2002). Mean age (our sample: 34 years; Patel: 36 years), median Glasgow Coma Score (GCS) on admission (our sample: 7; Patel: 7) and median Injury Severity Score (ISS) (Greenspan et al., 1985) (our sample: 20; Patel: 25) were broadly comparable. The surviving population at 6 months reported by Patel consisted of 3% with Glasgow Outcome Score (GOS) 2; 20% with GOS 3; 26% with GOS 4 and 49% with GOS 5. The results for our sample were: 0% with GOS 2 (unable to complete assessments); 13% with GOS 3; 45% with GOS 4 and 42% with GOS 5. This may suggest a slightly inflated participant response from the GOS 4 outcome group (see Discussion).
Neuropsychological assessment
The computerized tasks were taken from the Cambridge Neuropsychological Test Automated Battery (CANTAB) (www.camcog.com) and were run on an Advantech personal computer (Model PPC-120T-RT) and responses registered either via the touch sensitive screen or a response key, depending on the task. Participants were seated ∼0.5 m from the computer screen and asked to make responses by touching the screen with their dominant hand. The volunteers completed a series of tasks, which are detailed in Table 1.
Summary of CANTAB tasks used
| Task | Description | References | Outcome measure |
|---|---|---|---|
| Simple reaction time | Assesses simple reaction time | Owen et al., 1995 | Reaction time |
| Pattern recognition | A two-choice test of abstract visual pattern recognition memory | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Spatial recognition | Test of spatial recognition memory in a two-choice forced discrimination paradigm | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Paired associate learning | A test of the ability to form visuo-spatial associations | Owen et al., 1995 | Number of errors to criterion |
| Rapid visual information processing | A test of sustained attention to detect infrequent three-digit sequences from among serially presented digits | Park et al., 1994Sahakian et al., 1989 | A′ (a measure of ability to detect targets)B′ (a measure of tendency to respond regardless of whethertarget present)Mean latency for correcttargets (ms) |
| Spatial working memory | A test of spatial working memory and strategy performance to find individually hidden ‘blue tokens’ without returning to a box where one has previously been found. | Owen et al., 1990 | Strategy scoreTotal between errors (returning to a box where a token has been found)Total within errors (returning to a box that has already been inspected) |
| Set shifting | Discrimination learning, testing the ability to selectively attend to and set shift between stimulus dimensions | Downes et al., 1989 | Number of stages completed |
| Spatial span | A test of spatial memory span to recall the order in which a series of boxes were highlighted | Owen et al., 1990 | Span length |
| Task | Description | References | Outcome measure |
|---|---|---|---|
| Simple reaction time | Assesses simple reaction time | Owen et al., 1995 | Reaction time |
| Pattern recognition | A two-choice test of abstract visual pattern recognition memory | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Spatial recognition | Test of spatial recognition memory in a two-choice forced discrimination paradigm | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Paired associate learning | A test of the ability to form visuo-spatial associations | Owen et al., 1995 | Number of errors to criterion |
| Rapid visual information processing | A test of sustained attention to detect infrequent three-digit sequences from among serially presented digits | Park et al., 1994Sahakian et al., 1989 | A′ (a measure of ability to detect targets)B′ (a measure of tendency to respond regardless of whethertarget present)Mean latency for correcttargets (ms) |
| Spatial working memory | A test of spatial working memory and strategy performance to find individually hidden ‘blue tokens’ without returning to a box where one has previously been found. | Owen et al., 1990 | Strategy scoreTotal between errors (returning to a box where a token has been found)Total within errors (returning to a box that has already been inspected) |
| Set shifting | Discrimination learning, testing the ability to selectively attend to and set shift between stimulus dimensions | Downes et al., 1989 | Number of stages completed |
| Spatial span | A test of spatial memory span to recall the order in which a series of boxes were highlighted | Owen et al., 1990 | Span length |
Summary of CANTAB tasks used
| Task | Description | References | Outcome measure |
|---|---|---|---|
| Simple reaction time | Assesses simple reaction time | Owen et al., 1995 | Reaction time |
| Pattern recognition | A two-choice test of abstract visual pattern recognition memory | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Spatial recognition | Test of spatial recognition memory in a two-choice forced discrimination paradigm | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Paired associate learning | A test of the ability to form visuo-spatial associations | Owen et al., 1995 | Number of errors to criterion |
| Rapid visual information processing | A test of sustained attention to detect infrequent three-digit sequences from among serially presented digits | Park et al., 1994Sahakian et al., 1989 | A′ (a measure of ability to detect targets)B′ (a measure of tendency to respond regardless of whethertarget present)Mean latency for correcttargets (ms) |
| Spatial working memory | A test of spatial working memory and strategy performance to find individually hidden ‘blue tokens’ without returning to a box where one has previously been found. | Owen et al., 1990 | Strategy scoreTotal between errors (returning to a box where a token has been found)Total within errors (returning to a box that has already been inspected) |
| Set shifting | Discrimination learning, testing the ability to selectively attend to and set shift between stimulus dimensions | Downes et al., 1989 | Number of stages completed |
| Spatial span | A test of spatial memory span to recall the order in which a series of boxes were highlighted | Owen et al., 1990 | Span length |
| Task | Description | References | Outcome measure |
|---|---|---|---|
| Simple reaction time | Assesses simple reaction time | Owen et al., 1995 | Reaction time |
| Pattern recognition | A two-choice test of abstract visual pattern recognition memory | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Spatial recognition | Test of spatial recognition memory in a two-choice forced discrimination paradigm | Owen et al., 1995Sahakian et al., 1988 | Percentage correctReaction time |
| Paired associate learning | A test of the ability to form visuo-spatial associations | Owen et al., 1995 | Number of errors to criterion |
| Rapid visual information processing | A test of sustained attention to detect infrequent three-digit sequences from among serially presented digits | Park et al., 1994Sahakian et al., 1989 | A′ (a measure of ability to detect targets)B′ (a measure of tendency to respond regardless of whethertarget present)Mean latency for correcttargets (ms) |
| Spatial working memory | A test of spatial working memory and strategy performance to find individually hidden ‘blue tokens’ without returning to a box where one has previously been found. | Owen et al., 1990 | Strategy scoreTotal between errors (returning to a box where a token has been found)Total within errors (returning to a box that has already been inspected) |
| Set shifting | Discrimination learning, testing the ability to selectively attend to and set shift between stimulus dimensions | Downes et al., 1989 | Number of stages completed |
| Spatial span | A test of spatial memory span to recall the order in which a series of boxes were highlighted | Owen et al., 1990 | Span length |
Statistical analysis
Analyses were carried out using the Statistical Package for Social Sciences V11.0.1 [SPSS Inc, Chicago (IL), USA]. Non-parametric statistics (Mann–Whitney U-test) were used as the distribution of the data violated assumptions of normality. It should be noted that a number of statistical tests were used in the neuropsychological analysis and, as such, significant results may capitalise on chance and the overall probability of a Type I error may exceed 5%. However, the cholinergic hypothesis predicts a specific pattern of results across a range of tasks, including both impaired and intact performances. Setting the acceptable alpha too low would reduce the power of detecting a group difference on tasks for which intact performance is predicted. To lower the probability of capitalizing on chance, analyses were planned a priori and it is the pattern of results that was interpreted.
MRI
A subgroup of volunteers (see Results) underwent MRI scans performed on a Bruker Medspec 30/100 spectrometer (Bruker Medical, Etlington, Germany) attached to an Oxford 3.0 T, 910 mm bore whole body, actively shielded, magnet (Oxford Magnet Technology, Oxford, UK).
A multi-oblique three-dimensional spoiled gradient-echo (3D SPGR) sequence was acquired using a repetition time (TR) of 19 ms, an echo time (TE) of 5 ms, a flip angle of 25°, a field of view of 256 × 220 × 180 mm and a matrix size of 256 × 220 × 180 (giving a spatial resolution of 1 × 1 × 1 mm).
MR analysis
The 3D SPGR scans were analysed using Statistical Parametric Mapping (SPM2) software (Wellcome Department of Cognitive Neurology, University College London, London, UK). Each scan was pre-processed in accordance with the VBM protocol described by Good et al. (2001). Briefly, the data were normalized to a standardized template (Friston et al., 1995; Ashburner and Friston, 2000). The data were normalized by global grey matter; this is a fundamental component that accounts for any difference among participants that are simply due to differences in brain size. The images were segmented using a Bayesian algorithm (Ashburner et al., 1997) and continuous probability maps were produced where the values correspond to the posterior probability that the voxel belonged to the grey matter partition. White and grey matter segmented images were smoothed with 12 mm isotropic Gaussian kernels, which render the voxel values an index of the amount of grey (or white) matter per unit volume under the smoothing kernel.
Inferences from statistical parametric maps were made at the False Discovery Rate (FDR) statistical threshold level, which is corrected for multiple comparisons across the entire brain (Genovese et al., 2002).
Results
The head injury group consisted of seven females and 24 males, while the control group consisted of 16 females and 16 males. Both groups had MMSE (head injury group: 27.9 ± 0.3; control group: 28.9 ± 0.2) and NART (head injury group: 105.9 ± 2.0; control group: 110.7 ± 1.4) scores within the normal range. There were no significant differences in age (head injury group: 34 ± 2.8 years; control group: 35 ± 2.4 years), NART scores or gender for the head injury and control groups. There was a significant difference between the groups on the MMSE score (Mann–Whitney U-test: Z = −2.8, P = 0.005), although both groups were within the normal range. The patients' head injuries were caused by road traffic accidents (n = 13), falls (n = 10), assaults (n = 4) and cycle accidents (n = 2). The cause of the remaining two individuals' injuries was unknown. Six individuals in the head injury group suffered from epilepsy (19%). The clinical characteristics of the head injury group are shown in Table 2.
Clinical characteristics of patient group studied
| Patient no. | Age | Gender | GCS on admission | Marshall score | Days in intensive care unit | GOS | ISS | APACHE |
|---|---|---|---|---|---|---|---|---|
| 1 | 29 | M | 9 | NEML | 13 | 5 | 25 | 7 |
| 2 | 20 | F | 6 | DI II | 11 | 4 | 16 | 18 |
| 3 | 24 | F | 7 | EML | 17 | 4 | 25 | 20 |
| 4 | 28 | M | 12 | DI II | 12 | 5 | 9 | 3 |
| 5 | 23 | M | 12 | DI II | 17 | 3 | 25 | 16 |
| 6 | 55 | M | 10 | DI II | 10 | 4 | 16 | 6 |
| 7 | 59 | F | 4 | EML | 14 | 3 | 25 | 21 |
| 8 | 20 | F | 4 | EML | 20 | 5 | 16 | 13 |
| 9 | 24 | M | 8 | DI III | 11 | 5 | 16 | 12 |
| 10 | 69 | F | 8 | DI II | 15 | 5 | 17 | 14 |
| 11 | 55 | M | 10 | NEML | 22 | 5 | 16 | 14 |
| 12 | 40 | M | 10 | DI II | 6 | 4 | 9 | 5 |
| 13 | 44 | M | 9 | EML | 10 | 4 | 16 | 10 |
| 14 | 37 | F | 7 | DI II | 6 | 5 | 34 | 6 |
| 15 | 36 | M | 8 | EML | 14 | 4 | 9 | 8 |
| 16 | 41 | M | 6 | EML | 7 | 4 | 25 | 6 |
| 17 | 35 | M | 3 | DI II | 7 | 4 | 25 | 15 |
| 18 | 18 | M | 7 | DI III | 14 | 3 | 30 | 11 |
| 19 | 27 | M | 6 | EML | 27 | 4 | 16 | 16 |
| 20 | 27 | M | 3 | DI IV | 12 | 4 | 27 | 19 |
| 21 | 30 | F | 4 | DI II | 12 | 3 | 25 | 6 |
| 22 | 59 | F | 6 | DI II | 12 | 4 | 25 | 9 |
| 23 | 22 | M | 6 | DI II | 4 | 5 | 17 | 13 |
| 24 | 28 | M | 11 | DI II | 10 | 5 | 16 | 32 |
| 25 | 37 | M | 9 | NML | 16 | 5 | 17 | 14 |
| 26 | 29 | M | 5 | NEML | 7 | 5 | 21 | 6 |
| 27 | 28 | M | 7 | EML | 3 | 4 | 25 | 14 |
| 28 | 70 | M | 12 | DI II | 15 | 5 | 17 | 14 |
| 29 | 27 | M | 9 | DI II | 27 | 4 | 17 | 10 |
| 30 | 41 | M | 4 | DI II | 8 | 5 | 38 | 18 |
| 31 | 35 | M | 11 | NEML | 20 | 4 | 16 | 4 |
| Patient no. | Age | Gender | GCS on admission | Marshall score | Days in intensive care unit | GOS | ISS | APACHE |
|---|---|---|---|---|---|---|---|---|
| 1 | 29 | M | 9 | NEML | 13 | 5 | 25 | 7 |
| 2 | 20 | F | 6 | DI II | 11 | 4 | 16 | 18 |
| 3 | 24 | F | 7 | EML | 17 | 4 | 25 | 20 |
| 4 | 28 | M | 12 | DI II | 12 | 5 | 9 | 3 |
| 5 | 23 | M | 12 | DI II | 17 | 3 | 25 | 16 |
| 6 | 55 | M | 10 | DI II | 10 | 4 | 16 | 6 |
| 7 | 59 | F | 4 | EML | 14 | 3 | 25 | 21 |
| 8 | 20 | F | 4 | EML | 20 | 5 | 16 | 13 |
| 9 | 24 | M | 8 | DI III | 11 | 5 | 16 | 12 |
| 10 | 69 | F | 8 | DI II | 15 | 5 | 17 | 14 |
| 11 | 55 | M | 10 | NEML | 22 | 5 | 16 | 14 |
| 12 | 40 | M | 10 | DI II | 6 | 4 | 9 | 5 |
| 13 | 44 | M | 9 | EML | 10 | 4 | 16 | 10 |
| 14 | 37 | F | 7 | DI II | 6 | 5 | 34 | 6 |
| 15 | 36 | M | 8 | EML | 14 | 4 | 9 | 8 |
| 16 | 41 | M | 6 | EML | 7 | 4 | 25 | 6 |
| 17 | 35 | M | 3 | DI II | 7 | 4 | 25 | 15 |
| 18 | 18 | M | 7 | DI III | 14 | 3 | 30 | 11 |
| 19 | 27 | M | 6 | EML | 27 | 4 | 16 | 16 |
| 20 | 27 | M | 3 | DI IV | 12 | 4 | 27 | 19 |
| 21 | 30 | F | 4 | DI II | 12 | 3 | 25 | 6 |
| 22 | 59 | F | 6 | DI II | 12 | 4 | 25 | 9 |
| 23 | 22 | M | 6 | DI II | 4 | 5 | 17 | 13 |
| 24 | 28 | M | 11 | DI II | 10 | 5 | 16 | 32 |
| 25 | 37 | M | 9 | NML | 16 | 5 | 17 | 14 |
| 26 | 29 | M | 5 | NEML | 7 | 5 | 21 | 6 |
| 27 | 28 | M | 7 | EML | 3 | 4 | 25 | 14 |
| 28 | 70 | M | 12 | DI II | 15 | 5 | 17 | 14 |
| 29 | 27 | M | 9 | DI II | 27 | 4 | 17 | 10 |
| 30 | 41 | M | 4 | DI II | 8 | 5 | 38 | 18 |
| 31 | 35 | M | 11 | NEML | 20 | 4 | 16 | 4 |
Marshall score: classification based on computerized tomography (Marshall et al., 1991). Higher scores of GCS, GOS and ISS indicate greater severity (Greenspan et al., 1985). APACHE = acute physiology and chronic health evaluation (high scores indicate greater severity of disease) (Knaus et al., 1985); DI = diffuse injury; (N)EML = (non) evacuated mass lesion.
Clinical characteristics of patient group studied
| Patient no. | Age | Gender | GCS on admission | Marshall score | Days in intensive care unit | GOS | ISS | APACHE |
|---|---|---|---|---|---|---|---|---|
| 1 | 29 | M | 9 | NEML | 13 | 5 | 25 | 7 |
| 2 | 20 | F | 6 | DI II | 11 | 4 | 16 | 18 |
| 3 | 24 | F | 7 | EML | 17 | 4 | 25 | 20 |
| 4 | 28 | M | 12 | DI II | 12 | 5 | 9 | 3 |
| 5 | 23 | M | 12 | DI II | 17 | 3 | 25 | 16 |
| 6 | 55 | M | 10 | DI II | 10 | 4 | 16 | 6 |
| 7 | 59 | F | 4 | EML | 14 | 3 | 25 | 21 |
| 8 | 20 | F | 4 | EML | 20 | 5 | 16 | 13 |
| 9 | 24 | M | 8 | DI III | 11 | 5 | 16 | 12 |
| 10 | 69 | F | 8 | DI II | 15 | 5 | 17 | 14 |
| 11 | 55 | M | 10 | NEML | 22 | 5 | 16 | 14 |
| 12 | 40 | M | 10 | DI II | 6 | 4 | 9 | 5 |
| 13 | 44 | M | 9 | EML | 10 | 4 | 16 | 10 |
| 14 | 37 | F | 7 | DI II | 6 | 5 | 34 | 6 |
| 15 | 36 | M | 8 | EML | 14 | 4 | 9 | 8 |
| 16 | 41 | M | 6 | EML | 7 | 4 | 25 | 6 |
| 17 | 35 | M | 3 | DI II | 7 | 4 | 25 | 15 |
| 18 | 18 | M | 7 | DI III | 14 | 3 | 30 | 11 |
| 19 | 27 | M | 6 | EML | 27 | 4 | 16 | 16 |
| 20 | 27 | M | 3 | DI IV | 12 | 4 | 27 | 19 |
| 21 | 30 | F | 4 | DI II | 12 | 3 | 25 | 6 |
| 22 | 59 | F | 6 | DI II | 12 | 4 | 25 | 9 |
| 23 | 22 | M | 6 | DI II | 4 | 5 | 17 | 13 |
| 24 | 28 | M | 11 | DI II | 10 | 5 | 16 | 32 |
| 25 | 37 | M | 9 | NML | 16 | 5 | 17 | 14 |
| 26 | 29 | M | 5 | NEML | 7 | 5 | 21 | 6 |
| 27 | 28 | M | 7 | EML | 3 | 4 | 25 | 14 |
| 28 | 70 | M | 12 | DI II | 15 | 5 | 17 | 14 |
| 29 | 27 | M | 9 | DI II | 27 | 4 | 17 | 10 |
| 30 | 41 | M | 4 | DI II | 8 | 5 | 38 | 18 |
| 31 | 35 | M | 11 | NEML | 20 | 4 | 16 | 4 |
| Patient no. | Age | Gender | GCS on admission | Marshall score | Days in intensive care unit | GOS | ISS | APACHE |
|---|---|---|---|---|---|---|---|---|
| 1 | 29 | M | 9 | NEML | 13 | 5 | 25 | 7 |
| 2 | 20 | F | 6 | DI II | 11 | 4 | 16 | 18 |
| 3 | 24 | F | 7 | EML | 17 | 4 | 25 | 20 |
| 4 | 28 | M | 12 | DI II | 12 | 5 | 9 | 3 |
| 5 | 23 | M | 12 | DI II | 17 | 3 | 25 | 16 |
| 6 | 55 | M | 10 | DI II | 10 | 4 | 16 | 6 |
| 7 | 59 | F | 4 | EML | 14 | 3 | 25 | 21 |
| 8 | 20 | F | 4 | EML | 20 | 5 | 16 | 13 |
| 9 | 24 | M | 8 | DI III | 11 | 5 | 16 | 12 |
| 10 | 69 | F | 8 | DI II | 15 | 5 | 17 | 14 |
| 11 | 55 | M | 10 | NEML | 22 | 5 | 16 | 14 |
| 12 | 40 | M | 10 | DI II | 6 | 4 | 9 | 5 |
| 13 | 44 | M | 9 | EML | 10 | 4 | 16 | 10 |
| 14 | 37 | F | 7 | DI II | 6 | 5 | 34 | 6 |
| 15 | 36 | M | 8 | EML | 14 | 4 | 9 | 8 |
| 16 | 41 | M | 6 | EML | 7 | 4 | 25 | 6 |
| 17 | 35 | M | 3 | DI II | 7 | 4 | 25 | 15 |
| 18 | 18 | M | 7 | DI III | 14 | 3 | 30 | 11 |
| 19 | 27 | M | 6 | EML | 27 | 4 | 16 | 16 |
| 20 | 27 | M | 3 | DI IV | 12 | 4 | 27 | 19 |
| 21 | 30 | F | 4 | DI II | 12 | 3 | 25 | 6 |
| 22 | 59 | F | 6 | DI II | 12 | 4 | 25 | 9 |
| 23 | 22 | M | 6 | DI II | 4 | 5 | 17 | 13 |
| 24 | 28 | M | 11 | DI II | 10 | 5 | 16 | 32 |
| 25 | 37 | M | 9 | NML | 16 | 5 | 17 | 14 |
| 26 | 29 | M | 5 | NEML | 7 | 5 | 21 | 6 |
| 27 | 28 | M | 7 | EML | 3 | 4 | 25 | 14 |
| 28 | 70 | M | 12 | DI II | 15 | 5 | 17 | 14 |
| 29 | 27 | M | 9 | DI II | 27 | 4 | 17 | 10 |
| 30 | 41 | M | 4 | DI II | 8 | 5 | 38 | 18 |
| 31 | 35 | M | 11 | NEML | 20 | 4 | 16 | 4 |
Marshall score: classification based on computerized tomography (Marshall et al., 1991). Higher scores of GCS, GOS and ISS indicate greater severity (Greenspan et al., 1985). APACHE = acute physiology and chronic health evaluation (high scores indicate greater severity of disease) (Knaus et al., 1985); DI = diffuse injury; (N)EML = (non) evacuated mass lesion.
Neuropsychological assessment
As can be seen in Table 3, performance on several of the subtests of the CANTAB battery (namely spatial span, spatial working memory and set shifting) did not differ between the two groups. By contrast, there was a significant difference on rapid visual information processing, pattern and spatial recognition, reaction time and paired associate learning. The nature of these statistically significant differences is considered in detail below.
Summary of test results
| Head injured group | Control group | Z score (from Mann–Whitney U-test) | ||||
|---|---|---|---|---|---|---|
| Simple reaction time | ||||||
| Latency (ms) | 1046 ± 64 | 784 ± 36 | −3.5** | |||
| Pattern recognition | ||||||
| Percentage correct | 84.3 ± 1.7 | 90.0 ± 1.8 | −2.8** | |||
| Latency (ms) | 2517 ± 146 | 1879 ± 65 | −3.5** | |||
| Spatial recognition | ||||||
| Percentage correct | 79.7 ± 1.6 | 80.8 ± 1.8 | −0.7 | |||
| Latency (ms) | 2674 ± 233 | 2044 ± 106 | −2.4* | |||
| Paired associate learning | ||||||
| Errors at 6 box | 8.1 ± 2.3 | 3.2 ± 0.6 | −2.0* | |||
| Rapid visual information processing | ||||||
| A′ | 0.86 ± 0.01 | 0.90 ± 0.01 | −3.0** | |||
| B′ | 0.93 ± 0.01 | 0.90 ± 0.06 | −1.3 | |||
| Latency (ms) | 544 ± 31 | 475 ± 17 | −2.1* | |||
| Spatial working memory | ||||||
| Within errors | 2.2 ± 0.6 | 1.2 ± 0.3 | −1.4 | |||
| Between errors | 26.4 ± 3.0 | 20.1 ± 3.1 | −1.8 | |||
| Strategy | 32.8 ± 1.0 | 29.9 ± 1.5 | −1.5 | |||
| Set shifting | ||||||
| Stages completed | 8.4 ± 0.2 | 8.3 ± 0.2 | −0.8 | |||
| Spatial span | ||||||
| Span length | 5.9 ± 0.2 | 6.3 ± 0.3 | −0.8 | |||
| BDI | 8.9 ± 1.3 | 3.9 ± 0.6 | −3.0** | |||
| Head injured group | Control group | Z score (from Mann–Whitney U-test) | ||||
|---|---|---|---|---|---|---|
| Simple reaction time | ||||||
| Latency (ms) | 1046 ± 64 | 784 ± 36 | −3.5** | |||
| Pattern recognition | ||||||
| Percentage correct | 84.3 ± 1.7 | 90.0 ± 1.8 | −2.8** | |||
| Latency (ms) | 2517 ± 146 | 1879 ± 65 | −3.5** | |||
| Spatial recognition | ||||||
| Percentage correct | 79.7 ± 1.6 | 80.8 ± 1.8 | −0.7 | |||
| Latency (ms) | 2674 ± 233 | 2044 ± 106 | −2.4* | |||
| Paired associate learning | ||||||
| Errors at 6 box | 8.1 ± 2.3 | 3.2 ± 0.6 | −2.0* | |||
| Rapid visual information processing | ||||||
| A′ | 0.86 ± 0.01 | 0.90 ± 0.01 | −3.0** | |||
| B′ | 0.93 ± 0.01 | 0.90 ± 0.06 | −1.3 | |||
| Latency (ms) | 544 ± 31 | 475 ± 17 | −2.1* | |||
| Spatial working memory | ||||||
| Within errors | 2.2 ± 0.6 | 1.2 ± 0.3 | −1.4 | |||
| Between errors | 26.4 ± 3.0 | 20.1 ± 3.1 | −1.8 | |||
| Strategy | 32.8 ± 1.0 | 29.9 ± 1.5 | −1.5 | |||
| Set shifting | ||||||
| Stages completed | 8.4 ± 0.2 | 8.3 ± 0.2 | −0.8 | |||
| Spatial span | ||||||
| Span length | 5.9 ± 0.2 | 6.3 ± 0.3 | −0.8 | |||
| BDI | 8.9 ± 1.3 | 3.9 ± 0.6 | −3.0** | |||
Values shown for each variable are the mean and standard error mean for each group.
P < 0.05
P < 0.01.
Summary of test results
| Head injured group | Control group | Z score (from Mann–Whitney U-test) | ||||
|---|---|---|---|---|---|---|
| Simple reaction time | ||||||
| Latency (ms) | 1046 ± 64 | 784 ± 36 | −3.5** | |||
| Pattern recognition | ||||||
| Percentage correct | 84.3 ± 1.7 | 90.0 ± 1.8 | −2.8** | |||
| Latency (ms) | 2517 ± 146 | 1879 ± 65 | −3.5** | |||
| Spatial recognition | ||||||
| Percentage correct | 79.7 ± 1.6 | 80.8 ± 1.8 | −0.7 | |||
| Latency (ms) | 2674 ± 233 | 2044 ± 106 | −2.4* | |||
| Paired associate learning | ||||||
| Errors at 6 box | 8.1 ± 2.3 | 3.2 ± 0.6 | −2.0* | |||
| Rapid visual information processing | ||||||
| A′ | 0.86 ± 0.01 | 0.90 ± 0.01 | −3.0** | |||
| B′ | 0.93 ± 0.01 | 0.90 ± 0.06 | −1.3 | |||
| Latency (ms) | 544 ± 31 | 475 ± 17 | −2.1* | |||
| Spatial working memory | ||||||
| Within errors | 2.2 ± 0.6 | 1.2 ± 0.3 | −1.4 | |||
| Between errors | 26.4 ± 3.0 | 20.1 ± 3.1 | −1.8 | |||
| Strategy | 32.8 ± 1.0 | 29.9 ± 1.5 | −1.5 | |||
| Set shifting | ||||||
| Stages completed | 8.4 ± 0.2 | 8.3 ± 0.2 | −0.8 | |||
| Spatial span | ||||||
| Span length | 5.9 ± 0.2 | 6.3 ± 0.3 | −0.8 | |||
| BDI | 8.9 ± 1.3 | 3.9 ± 0.6 | −3.0** | |||
| Head injured group | Control group | Z score (from Mann–Whitney U-test) | ||||
|---|---|---|---|---|---|---|
| Simple reaction time | ||||||
| Latency (ms) | 1046 ± 64 | 784 ± 36 | −3.5** | |||
| Pattern recognition | ||||||
| Percentage correct | 84.3 ± 1.7 | 90.0 ± 1.8 | −2.8** | |||
| Latency (ms) | 2517 ± 146 | 1879 ± 65 | −3.5** | |||
| Spatial recognition | ||||||
| Percentage correct | 79.7 ± 1.6 | 80.8 ± 1.8 | −0.7 | |||
| Latency (ms) | 2674 ± 233 | 2044 ± 106 | −2.4* | |||
| Paired associate learning | ||||||
| Errors at 6 box | 8.1 ± 2.3 | 3.2 ± 0.6 | −2.0* | |||
| Rapid visual information processing | ||||||
| A′ | 0.86 ± 0.01 | 0.90 ± 0.01 | −3.0** | |||
| B′ | 0.93 ± 0.01 | 0.90 ± 0.06 | −1.3 | |||
| Latency (ms) | 544 ± 31 | 475 ± 17 | −2.1* | |||
| Spatial working memory | ||||||
| Within errors | 2.2 ± 0.6 | 1.2 ± 0.3 | −1.4 | |||
| Between errors | 26.4 ± 3.0 | 20.1 ± 3.1 | −1.8 | |||
| Strategy | 32.8 ± 1.0 | 29.9 ± 1.5 | −1.5 | |||
| Set shifting | ||||||
| Stages completed | 8.4 ± 0.2 | 8.3 ± 0.2 | −0.8 | |||
| Spatial span | ||||||
| Span length | 5.9 ± 0.2 | 6.3 ± 0.3 | −0.8 | |||
| BDI | 8.9 ± 1.3 | 3.9 ± 0.6 | −3.0** | |||
Values shown for each variable are the mean and standard error mean for each group.
P < 0.05
P < 0.01.
Simple reaction time
The head injured group had a significantly slower reaction time than controls (Z = −3.5, P < 0.001).
Pattern recognition
The head injured group made significantly more errors in pattern recognition than controls (Z = −2.7, P = 0.006) and responded much more slowly than controls (Z = −3.5, P < 0.001).
Spatial recognition
The accuracy of the two groups at spatial recognition did not differ significantly (Z = −0.7, P > 0.4). However the head injured group had significantly longer response times (Z = −2.4, P = 0.015).
Paired associate learning
The head injured group made significantly more errors at this task than the control group (Z = −1.9, P = 0.049).
Rapid visual information processing
The two groups performed similarly on the measure of B′(Z = −1.3, P > 0.19). However the head injured group had significantly longer response times (Z = −2.0, P = 0.038), which could be accounted for using the results from the simple reaction time task. The head injured group scored significantly lower on A′; indicative of reduced levels of target detection (Z = −3.0, P = 0.003).
Depression
The BDI scores were significantly higher in the head injured group compared with the controls (Z = −3.3, P = 0.001). In order to determine whether the higher incidence of depression influenced the neuropsychological results, the statistical analysis was carried out excluding all individuals (n = 13) scoring in the range of clinically significant depression (BDI >10). The pattern of results remained the same as reported above, with the exception of the Paired Associate Learning test, where there was no significant difference in accuracy (Z = −1.1, P > 0.2).
MRI
Nine individuals out of the whole cohort of 31 patients in the head injured group did not complete an MRI scan either due to metallic implants or claustrophobia. Consequently, 22 patients were scanned (seven females, 15 males). The number of individuals in the control scanned group was selected to match the number in the head injured group (12 females, 11 males). The neuropsychological profiles of the scanned subgroups did not differ from the overall group profile.
The VBM analysis revealed a diffuse pattern of abnormality, which is detailed in Tables 4 and 5. Significant decreases in grey matter density were found in the region of the acetylcholinergic basal forebrain nuclei and in areas receiving projections from all of the major cholinergic pathways (such as bilateral hippocampal formation, bilateral insula and the thalamus). Significant decreases in white matter density were found in the lateral capsular pathway and the medial pathway.
Areas of significant decreases in grey matter density in the head injured group compared with controls
| Location | Coordinates (x, y, z) in mm | Z score | FDR corresponding P value | |||
|---|---|---|---|---|---|---|
| ACh nuclei | ||||||
| Basal forebrain (including septal nuclei; diagonal band of Broca; nucleus basalis of Meynert) | −2 4 2 | 5.83 | <0.001 | |||
| Dorsal tegmental area | 6 −29 −4 | 6.04 | <0.001 | |||
| Septal pathway projections | ||||||
| Left hippocampal formation | −21 −11 −22 | 4.48 | <0.001 | |||
| Right hippocampal formation | 20 −11 −23 | 3.45 | 0.007 | |||
| Lateral perisylvian pathway projections | ||||||
| Left insula* | −36 16 2 | 4.52 | <0.001 | |||
| Right insula* | 55 −12 10 | 4.48 | <0.001 | |||
| Lateral capsular pathway projections | ||||||
| Left parietal lobe* | −55 −40 45 | 3.34 | 0.010 | |||
| Left occipital lobe | −26 −83 2 | 3.48 | 0.007 | |||
| Right temporal lobe* | 36 13 −28 | 4.37 | <0.001 | |||
| Dorsal tegmental projections | ||||||
| Thalamus | 0 −11 14 | 5.81 | <0.001 | |||
| Cerebellum* | −10 −42 −29 | 3.34 | 0.010 | |||
| Location | Coordinates (x, y, z) in mm | Z score | FDR corresponding P value | |||
|---|---|---|---|---|---|---|
| ACh nuclei | ||||||
| Basal forebrain (including septal nuclei; diagonal band of Broca; nucleus basalis of Meynert) | −2 4 2 | 5.83 | <0.001 | |||
| Dorsal tegmental area | 6 −29 −4 | 6.04 | <0.001 | |||
| Septal pathway projections | ||||||
| Left hippocampal formation | −21 −11 −22 | 4.48 | <0.001 | |||
| Right hippocampal formation | 20 −11 −23 | 3.45 | 0.007 | |||
| Lateral perisylvian pathway projections | ||||||
| Left insula* | −36 16 2 | 4.52 | <0.001 | |||
| Right insula* | 55 −12 10 | 4.48 | <0.001 | |||
| Lateral capsular pathway projections | ||||||
| Left parietal lobe* | −55 −40 45 | 3.34 | 0.010 | |||
| Left occipital lobe | −26 −83 2 | 3.48 | 0.007 | |||
| Right temporal lobe* | 36 13 −28 | 4.37 | <0.001 | |||
| Dorsal tegmental projections | ||||||
| Thalamus | 0 −11 14 | 5.81 | <0.001 | |||
| Cerebellum* | −10 −42 −29 | 3.34 | 0.010 | |||
Additional peaks in same anatomical regions not listed.
Areas of significant decreases in grey matter density in the head injured group compared with controls
| Location | Coordinates (x, y, z) in mm | Z score | FDR corresponding P value | |||
|---|---|---|---|---|---|---|
| ACh nuclei | ||||||
| Basal forebrain (including septal nuclei; diagonal band of Broca; nucleus basalis of Meynert) | −2 4 2 | 5.83 | <0.001 | |||
| Dorsal tegmental area | 6 −29 −4 | 6.04 | <0.001 | |||
| Septal pathway projections | ||||||
| Left hippocampal formation | −21 −11 −22 | 4.48 | <0.001 | |||
| Right hippocampal formation | 20 −11 −23 | 3.45 | 0.007 | |||
| Lateral perisylvian pathway projections | ||||||
| Left insula* | −36 16 2 | 4.52 | <0.001 | |||
| Right insula* | 55 −12 10 | 4.48 | <0.001 | |||
| Lateral capsular pathway projections | ||||||
| Left parietal lobe* | −55 −40 45 | 3.34 | 0.010 | |||
| Left occipital lobe | −26 −83 2 | 3.48 | 0.007 | |||
| Right temporal lobe* | 36 13 −28 | 4.37 | <0.001 | |||
| Dorsal tegmental projections | ||||||
| Thalamus | 0 −11 14 | 5.81 | <0.001 | |||
| Cerebellum* | −10 −42 −29 | 3.34 | 0.010 | |||
| Location | Coordinates (x, y, z) in mm | Z score | FDR corresponding P value | |||
|---|---|---|---|---|---|---|
| ACh nuclei | ||||||
| Basal forebrain (including septal nuclei; diagonal band of Broca; nucleus basalis of Meynert) | −2 4 2 | 5.83 | <0.001 | |||
| Dorsal tegmental area | 6 −29 −4 | 6.04 | <0.001 | |||
| Septal pathway projections | ||||||
| Left hippocampal formation | −21 −11 −22 | 4.48 | <0.001 | |||
| Right hippocampal formation | 20 −11 −23 | 3.45 | 0.007 | |||
| Lateral perisylvian pathway projections | ||||||
| Left insula* | −36 16 2 | 4.52 | <0.001 | |||
| Right insula* | 55 −12 10 | 4.48 | <0.001 | |||
| Lateral capsular pathway projections | ||||||
| Left parietal lobe* | −55 −40 45 | 3.34 | 0.010 | |||
| Left occipital lobe | −26 −83 2 | 3.48 | 0.007 | |||
| Right temporal lobe* | 36 13 −28 | 4.37 | <0.001 | |||
| Dorsal tegmental projections | ||||||
| Thalamus | 0 −11 14 | 5.81 | <0.001 | |||
| Cerebellum* | −10 −42 −29 | 3.34 | 0.010 | |||
Additional peaks in same anatomical regions not listed.
Areas of significant decreases in white matter density in the head injured group compared with controls
| Location | Montreal Neurological Institute coordinates of peak | Z score | P value |
|---|---|---|---|
| Lateral capsular pathway | −33 −31 −3 | 4.37 | 0.020 |
| 28 −29 13 | 3.61 | 0.030 | |
| 40 27 −1 | 3.47 | 0.037 | |
| 24 24 25 | 3.31 | 0.047 | |
| Medial pathway | −1 24 4 | 3.59 | 0.031 |
| −8 −41 21 | 3.36 | 0.044 |
| Location | Montreal Neurological Institute coordinates of peak | Z score | P value |
|---|---|---|---|
| Lateral capsular pathway | −33 −31 −3 | 4.37 | 0.020 |
| 28 −29 13 | 3.61 | 0.030 | |
| 40 27 −1 | 3.47 | 0.037 | |
| 24 24 25 | 3.31 | 0.047 | |
| Medial pathway | −1 24 4 | 3.59 | 0.031 |
| −8 −41 21 | 3.36 | 0.044 |
Areas of significant decreases in white matter density in the head injured group compared with controls
| Location | Montreal Neurological Institute coordinates of peak | Z score | P value |
|---|---|---|---|
| Lateral capsular pathway | −33 −31 −3 | 4.37 | 0.020 |
| 28 −29 13 | 3.61 | 0.030 | |
| 40 27 −1 | 3.47 | 0.037 | |
| 24 24 25 | 3.31 | 0.047 | |
| Medial pathway | −1 24 4 | 3.59 | 0.031 |
| −8 −41 21 | 3.36 | 0.044 |
| Location | Montreal Neurological Institute coordinates of peak | Z score | P value |
|---|---|---|---|
| Lateral capsular pathway | −33 −31 −3 | 4.37 | 0.020 |
| 28 −29 13 | 3.61 | 0.030 | |
| 40 27 −1 | 3.47 | 0.037 | |
| 24 24 25 | 3.31 | 0.047 | |
| Medial pathway | −1 24 4 | 3.59 | 0.031 |
| −8 −41 21 | 3.36 | 0.044 |
Discussion
This study reports a neuropsychological profile and a pattern of structural abnormalities in a group of head injured patients consistent with a chronic cholinergic deficit. The head injured survivors had significant deficits in sustained attention, paired associate learning and reaction time, consistent with previous findings (Arcia and Gualtieri, 1994; Ruff et al., 1994; Whyte et al., 1995; Robertson et al., 1997; Hellawell et al., 1999; Zahn and Mirsky, 1999; Chan, 2000; Polo et al., 2002). These cognitive skills have been shown to depend on the integrity of the cholinergic system (Crow and Grove-White, 1973; Ridley et al., 1984; Muir et al., 1994; Voytko et al., 1994; Ragozzino et al., 1996; Gill et al., 2000; Himmelheber et al., 2000; Rezvani and Levin, 2001; Rogers and Kesner, 2003). In contrast, this profile is inconsistent with abnormalities of either the dopamine or the serotonin systems (e.g. Park et al., 1994; Harmer et al., 2001). While performance on these tasks is also modulated by pharmacological manipulation of the noradrenergic system (e.g. Jakala et al., 1999b; Middleton et al., 1999; Smith et al., 1999), spatial working memory performance is also affected by modulation of noradrenaline (Jakala et al., 1999a; Middleton et al., 1999). As the head injury group were unimpaired on the spatial working memory task, this suggests that noradrenaline abnormalities cannot per se account for the neuropsychological profile found in this study. The overall neuropsychological profile of the head injured patients is therefore consistent with cholinergic abnormality and indeed is very similar to that seen in patients with mild Alzheimer's disease (Sahakian et al., 1988).
Attributing the VBM results to particular neurochemical systems is difficult due to the overlapping projection patterns of the systems (see Table 6). In addition, the possibility that the abnormalities detected are not related to a particular neurotransmitter system should also be considered. However, the pattern of structural abnormality detected using VBM appears to be highly coincident with the distribution of the cholinergic system in the brain. In particular, areas known to be rich in cholinergic innervations (e.g. bilateral hippocampal formation and the thalamus (see Perry et al., 1999)) were found to have reduced grey matter density. Further, several areas of the cholinergic pathways described by Selden et al. (1998) had significant reductions in white matter density. Indeed, a considerable proportion of the cholinergic sources, projections and pathways in the human brain were found to be abnormal. Of the remaining cholinergic areas, some are likely to be structures beyond the resolution of the VBM methodology (e.g. the olfactory bulb) and other areas were encompassed by large clusters with maximal peaks elsewhere (e.g. see medial frontal abnormality in Fig. 2A).
Location of principal sources and projections of neurotransmitters
| Hypothalamus | Thalamus | Caudate | Putamen | Nucleus Accumbens | Globus Pallidus | Amgydala | Hippocampus | Olfactory bulb | Cerebellum | Pons | Medulla | Neocortex | Substantia nigra | Ventral tegmental area | Dorsal tegmental area | Nucleus of diagonal band | Septum | Nucleus basalis of Meynert | Locus Ceruleus | Raphe nucleus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DA | SP | S | P | P | P | P | P | P | P | P | S | S | P | P | P | ||||||
| NA | P | P | P | P | P | SP | P | P | S | ||||||||||||
| 5HT | P | P | P | P | P | P | P | P | P | P | P | P | P | S | |||||||
| ACh | P | P | P | P | P | P | S | S | S | S |
| Hypothalamus | Thalamus | Caudate | Putamen | Nucleus Accumbens | Globus Pallidus | Amgydala | Hippocampus | Olfactory bulb | Cerebellum | Pons | Medulla | Neocortex | Substantia nigra | Ventral tegmental area | Dorsal tegmental area | Nucleus of diagonal band | Septum | Nucleus basalis of Meynert | Locus Ceruleus | Raphe nucleus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DA | SP | S | P | P | P | P | P | P | P | P | S | S | P | P | P | ||||||
| NA | P | P | P | P | P | SP | P | P | S | ||||||||||||
| 5HT | P | P | P | P | P | P | P | P | P | P | P | P | P | S | |||||||
| ACh | P | P | P | P | P | P | S | S | S | S |
See: Cooper et al., 1991; Feldman et al., 1997; Hanaway et al., 1998; Selden et al., 1998; Perry et al., 1999. DA = dopamine; 5HT = serotonin; NA = noradrenaline; P = projections; S = source of neurotransmitters.
Location of principal sources and projections of neurotransmitters
| Hypothalamus | Thalamus | Caudate | Putamen | Nucleus Accumbens | Globus Pallidus | Amgydala | Hippocampus | Olfactory bulb | Cerebellum | Pons | Medulla | Neocortex | Substantia nigra | Ventral tegmental area | Dorsal tegmental area | Nucleus of diagonal band | Septum | Nucleus basalis of Meynert | Locus Ceruleus | Raphe nucleus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DA | SP | S | P | P | P | P | P | P | P | P | S | S | P | P | P | ||||||
| NA | P | P | P | P | P | SP | P | P | S | ||||||||||||
| 5HT | P | P | P | P | P | P | P | P | P | P | P | P | P | S | |||||||
| ACh | P | P | P | P | P | P | S | S | S | S |
| Hypothalamus | Thalamus | Caudate | Putamen | Nucleus Accumbens | Globus Pallidus | Amgydala | Hippocampus | Olfactory bulb | Cerebellum | Pons | Medulla | Neocortex | Substantia nigra | Ventral tegmental area | Dorsal tegmental area | Nucleus of diagonal band | Septum | Nucleus basalis of Meynert | Locus Ceruleus | Raphe nucleus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DA | SP | S | P | P | P | P | P | P | P | P | S | S | P | P | P | ||||||
| NA | P | P | P | P | P | SP | P | P | S | ||||||||||||
| 5HT | P | P | P | P | P | P | P | P | P | P | P | P | P | S | |||||||
| ACh | P | P | P | P | P | P | S | S | S | S |
See: Cooper et al., 1991; Feldman et al., 1997; Hanaway et al., 1998; Selden et al., 1998; Perry et al., 1999. DA = dopamine; 5HT = serotonin; NA = noradrenaline; P = projections; S = source of neurotransmitters.
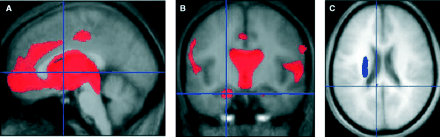
Representative results from VBM analysis. Red indicates areas of decreased grey matter density. Blue indicates areas of decreased white matter density. Images thresholded at FDR P < 0.01 (grey matter) and FDR P < 0.05 (white matter) (A) septal nuclei, diagonal band of Broca, nucleus basalis of Meynert (−2 4 2), (B) bilateral hippocampal formation (−21 −11 −22) and (C) medial pathway (−8 −41 21) (lateral capsular pathway abnormality also visible).
Given the diffuse nature of the abnormalities, it might be suggested that the pattern of abnormality is consistent with the distribution pattern of other neurotransmitter systems, for example dopamine or serotonin. However, the absence of detectable abnormality in the basal ganglia, rich in dopamine and serotonin (Kandel et al., 1991) combined with no abnormality being detected in source regions of dopamine, noradrenaline or serotonin (compared with ACh) suggest this hypothesis is unlikely (see Table 6). Further, the neuropsychological profile is also indicative of a cholinergic abnormality.
These results suggest that the cognitive sequelae associated with head injury may be ameliorated by drugs that enhance cholinergic function. Although chronic use of acetylcholinesterase inhibitors is now routine in the treatment of Alzheimer's disease (Rogers et al., 1998), use in head injury survivors is not current practice. The data from this study, combined with the case reports of cognitive improvements following cholinergic enhancement (Goldberg et al., 1982; Levin et al., 1986; Weinberg et al., 1987), suggest that large-scale, blinded drug trials are now warranted.
This study provides neuropsychological and structural imaging evidence that there is a chronic cholinergic deficit in head injury survivors. Future studies are necessary to confirm these findings and further elucidate the nature of the cholinergic dysfunction. In particular, use of recently developed PET ligands that bind to ACh receptors (e.g. 4N 11C methylpiperidylbenzilate (Sihver et al., 2001) and 123I quinuclidinyl benzilate (Kemp et al., 2003)) is likely to prove fruitful.
Data from post-mortem studies post fatal head injury support the hypothesis that the pattern of abnormality found in the VBM analysis of this cohort is linked to the cholinergic system (Dewar and Graham, 1996; Murdoch et al., 1998, 2002). It is possible, however, that such patterns obtained from fatal head injury may not be seen in individuals surviving head injury. This possibility is underlined by the heterogeneity found in post-mortem studies of survivors of head injury who later die of other causes. These studies reveal that a mixture of diffuse and focal injuries is present in individuals who survive with severe disability, moderate disability or good outcome (Blumbergs et al., 1994; Adams et al., 2001; Jennett et al., 2001). Indeed, it is notable that these studies report a very low incidence of thalamic abnormality and considerable heterogeneity in the samples.
The VBM data from our study are not fully consistent with the neuropathological appearances seen in previous studies. These discrepancies are likely to reflect the inclusion of individuals with alcoholism in other studies (Adams et al., 2001), although differing age ranges (Jennett et al., 2001), higher incidence of epilepsy (Adams et al., 2001) and small sample size (Blumbergs et al., 1994) may also account for differences between studies. Additionally, as acknowledged in Jennett et al. (2001), these studies recorded the presence or absence of lesions. More quantitative measures, particularly focussing on cholinergic markers, may reveal abnormalities consistent with the pattern found in this study. Our study population is generally representative of patients in whom neuropsychological testing is possible and appropriate; although they may not fully represent the spectrum of outcomes following head injury, they probably represent the prime therapeutic target for cognitive enhancement therapy. However, it is desirable that future studies should explore the relationship of cholinergic neuronal loss to changes on VBM and attempt to explain discrepancies between studies.
Survivors of head injury may have a higher risk of developing Alzheimer's disease (Heyman et al., 1984; French et al., 1985; Mortimer et al., 1985; Graves et al., 1990; Rasmusson et al., 1995; Nemetz et al., 1999; Lye and Shores, 2000; Fleminger et al., 2003; but see Shalat et al., 1987; Broe et al., 1990) and have been reported to have an earlier onset (Nemetz et al., 1999). The underlying mechanism for this vulnerability is unknown. However, chronic cholinergic deficits (as suggested by this study) would be consistent with this predisposition. Indeed the cognitive profile of the survivors falls midway between that of individuals with Alzheimer's disease and those with questionable dementia (see Swainson et al., 2001). It will be important to determine, in future work, whether there is any evidence that the cognitive abnormalities noted in this study are progressive, particularly as the Paired Associate Learning test has been found to be sensitive to the early detection of Alzheimer's disease (Blackwell et al., 2004; Swainson et al., 2001).
The mechanism underlying the disruption of the cholinergic system is unclear. Several different possibilities have been suggested in the literature. Cholinergic damage may occur as a result of ischaemia, as seen in microsphere embolized rats (Takagi et al., 1997). Consistent with the neuropathological pattern found in this study, the hippocampal formation is known to be exquisitely sensitive to ischaemia (Graham et al., 2002). However, Murdoch et al. (1998) found no correlation between ischaemia and abnormal CHAT levels, suggesting that ischaemia is unlikely to be the sole cause. There is some evidence to support a role for chronic hypoperfusion (de la Torre, 2000), while raised intracranial pressure (leading to internal herniation of the midline structures) may also be implicated in the basal forebrain abnormalities. Future studies should explore the relationship between the neurocognitive and structural abnormalities seen post head injury and both primary injury and secondary insults.
The finding of common structural abnormalities in the head injured group despite the hetereogeneous cause of injury (see above) suggests that the causal factors are likely to be secondary to the acute injury. Diffuse head injury caused by acceleration/deceleration leads to stereotyped injury to the inferior frontal and anteriotemporal poles of the brain. Evidence that cholinergic disruption is associated with secondary insults includes a negative correlation between survival time and CHAT levels in post-mortem studies (Murdoch et al., 1998). This is further supported by animal studies that reveal that reductions in ACh receptors noted at 24 h post injury are not detectable 3 h post injury (DeAngelis et al., 1994). The anatomical pattern of degeneration post head injury in animal models has also been shown to evolve over time, with only hippocampal abnormalities being detectable at first, followed later by thalamus and basal forebrain abnormalities (Leonard et al., 1997; Chen et al., 2003).
A number of researchers have suggested that head injury may lead to a selective disruption in the ACh pathways (e.g. synthesis, reuptake and degradation) (see Dixon et al., 1994b; Schmidt and Grady, 1995). For example, it has been suggested that the cholinergic neurons have a unique metabolic capacity, which renders them particularly vulnerable to damage. These neurons not only use choline to form the neurotransmitter acetylcholine, but also use choline in cell membrane synthesis. When choline is depleted [e.g. after the excitotoxic release of neurotransmitters which occurs post head injury (e.g. Hutchinson et al., 2002)], the neurons may use the choline bound in the cell membrane to create ACh. Such autocannibalism would lead to selective cholinergic cell loss (Wurtman, 1992).
Although the results from this study are consistent with a relatively selective deficit in the cholinergic system, it is important to acknowledge that this is unlikely to occur in isolation. Acetylcholine interacts with many other transmitters including dopamine, noradrenaline, serotonin, GABA and glutamate (see Lucas-Meunier et al., 2003) and cognitive functions are unlikely to be a simple function of just ACh. For example, in animal models, deficits associated with cholinergic disruption are further exacerbated by noradrenaline depletion (Decker and Gallagher, 1987). While gross disorder of neurotransmitter systems (other than the cholinergic system) is not consistent with the neuropsychological profile of the head injured individuals (see above), it is likely that mild abnormalities in the other neurotransmitter systems are present in the head injury group at a level where functional and structural abnormalities are not detected by neuropsychological assessment and MRI scans.
Limitations of the study
In order to perform VBM, it is necessary to co-register all the scans in stereotaxic space. This is done by using an automated algorithm that minimizes a measure of difference between the image and a template. It is possible that the normalization accuracy in the head injured group was compromised by the presence of focal lesions in some of the patients. We attempted to minimize these errors by not using non-linear transformations, which are especially vulnerable to inappropriate distortions (Brett et al., 2001). As we were interested in determining the location of structural abnormality, cost function masking was not used in order to avoid introducing bias for lesion location. Visual inspection confirmed that the normalized images did not contain distortions. While it is possible that imperfect spatial normalization may have affected the results given the heterogeneity of the injury causes and focal lesion location, we consider it unlikely that such differences would give rise to systematic differences.
The sample of patients in this study may not be representative of all head injury survivors (see Results). The study required participants to be able to comprehend simple instructions and cooperate with the testing session. However, in our experience, very few individuals are unable to meet these criteria, consistent with findings in other patient groups (e.g. Swainson et al., 2001). Indeed the mean MMSE score of ∼28 was close to ceiling (30) and well above the dementing range of <24. It is possible that survivors with good outcome are less inclined to contribute to research, particularly if they have already returned to work. Nevertheless, this study shows that at least a subgroup of head injured survivors may have chronic cholinergic dysfunction.
The high incidence of depression (13 out of 31) in our sample is comparable with levels reported in other studies (e.g. Mayou and Bryant, 2001). Although clinical depression is associated with cognitive dysfunction (e.g. Sweeney et al., 2000; Tavares et al., 2003), it is unlikely that cognitive deficits found in the head injured group can be attributed to the incidence of depression. First, the pattern of results remains very similar when the individuals with depression are excluded. Secondly, depression is associated with a broad range of deficits including tests of executive function (Elliott et al., 1997). Patients scoring within the clinical range on the BDI were not excluded from the overall study to maintain the representative nature of the sample of head injury survivors.
We wish to thank the patients and volunteers who participated in this study and the staff and radiographers at the Wolfson Brain Imaging Centre. This work was supported by the Medical Research Council (Grant number: G9439390 ID 56883) and the Fund for Addenbrooke's. It was carried out within the MRC Centre for Behavioural and Clinical Neuroscience.
References
Abe K, Inokawa M, Kashiwagi A, Yanagihara T. Amnesia after a discrete basal forebrain lesion.
Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage.
Arcia E, Gualtieri CT. Neurobehavioural performance of adults with closed-head injury, adults with attention deficit, and controls.
Arciniegas DB. The cholinergic hypothesis of cognitive impairment caused by traumatic brain injury.
Arciniegas D, Adler L, Topkoff J, Cawthra E, Filley CM, Reite M. Attention and memory dysfunction after traumatic brain injury: cholinergic mechanisms, sensory gating, and a hypothesis for further investigation.
Ashburner J, Neelin P, Collins DL, Evans A, Friston K. Incorporating prior knowledge into image registration.
Barefoot HC, Baker HF, Ridley RM. Synergistic effects of unilateral immunolesions of the cholinergic projections from the basal forebrain and contralateral ablations of the inferotemporal cortex and.
Bentivoglio M, Steriade M. Brainstem-diencephalic circuits as a structural substrate of the ascending reticular activation concept. In: Mancia M and Marini G, editors. The diencephalon and sleep. New York: Raven Press;
Blackwell AD, Sahakian BJ, Vesey R, Semple JM, Robbins TW, Hodges JR. Detecting dementia: novel neuropsychological markers of preclinical Alzheimer's disease.
Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Staining of amyloid precursor protein to study axonal damage in mild head injury.
Brett M, Leff AP, Rorden C, Ashburner J. Spatial normalization of brain images with focal lesions using cost function masking.
Broe GA, Henderson AS, Creasey H, McCusker E, Korten AE, Jorm AF, et al. A case-control study of Alzheimer's disease in Australia.
Chan RC. Attentional deficits in patients with closed head injury: a further study to the discriminative validity of the test of everyday attention.
Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury.
Cooper JR, Bloom FE, Roth RH. The biochemical basis of neuropharmacology. New York: Oxford University Press;
Crow TJ, Grove-White IG. An analysis of the learning deficit following hyoscine administration to man.
Dalley JW, McGaughy J, O'Connell MT, Cardinal RN, Levita L, Robbins TW. Distinct changes in cortical acetylcholine and noradrenaline efflux during contingent and noncontingent performance of a visual attentional task.
Damasio AR, Graff-Radford NR, Eslinger PJ, Damasio H, Kassell N. Amnesia following basal forebrain lesions.
DeAngelis MM, Hayes RL, Lyeth BG. Traumatic brain injury causes a decrease in M2 muscarinic cholinergic receptor binding in the rat brain.
Decker MW, Gallagher M. Scopolamine-disruption of radial arm maze performance: modification by noradrenergic depletion.
Delahunty TM. Mild traumatic brain injury enhances muscarinic receptor-linked inositol phosphate production in rat hippocampus.
Delahunty TM, Jiang JY, Lyeth BG. Role for muscarinic and metabotropic receptors in brain trauma.
de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer's pathogenesis.
Dewar D, Graham DI. Depletion of choline acetyltransferase activity but preservation of M1 and M2 muscarinic receptor binding sites in temporal cortex following head injury: a preliminary.
Dixon CE, Bao J, Bergmann JS, Johnson KM. Traumatic brain injury reduces hippocampal high-affinity [3H]choline uptake but not extracellular choline levels in rats.
Dixon CE, Hamm RJ, Taft WC, Hayes RL. Increased anticholinergic sensitivity following closed skull impact and controlled cortical impact traumatic brain injury in the rat.
Dixon CE, Bao J, Johnson KM, Yang K, Whitson J, Clifton GL, et al. Basal and scopolamine-evoked release of hippocampal acetylcholine following traumatic brain injury in rats.
Dixon CE, Flinn P, Bao J, Venya R, Hayes RL. Nerve growth factor attenuates cholinergic deficits following traumatic brain injury in rats.
Dixon CE, Ma X, Marion DW. Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury.
Downes JJ, Roberts AC, Sahakian BJ, Evenden JL, Morris RG, Robbins TW. Impaired extra-dimensional shift performance in medicated and unmedicated Parkinson's disease: evidence for a specific attentional dysfunction.
Elliott R, Baker SC, Rogers RD, O'Leary DA, Paykel ES, Frith CD, et al. Prefrontal dysfunction in depressed patients performing a complex planning task: a study using positron emission tomography.
Feldman RS, Meyer J, S, Quenzer LF. Principles of neuropsychopharmacology. Massachusetts: Sinauer Associates;
Field JH. Epidemiology in head injury in England and Wales. London: Research Division, Department of Health and Social Security;
Fine A, Hoyle C, Maclean CJ, Levatte TL, Baker HF, Ridley RM. Learning impairments following injection of a selective cholinergic immunotoxin, ME20.4 IgG-saporin, into the basal nucleus of Meynert in monkeys.
Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication.
Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician.
Fortin S, Godbout L, Braun CM. Cognitive structure of executive deficits in frontally lesioned head trauma patients performing activities of daily living.
French LR, Schuman LM, Mortimer JA, Hutton JT, Boatman RA, Christians B. A case-control study of dementia of the Alzheimer type.
Friston KJ, Ashburner J, Frith CD, Poline JB, Heather JD, Frackowiak RSJ. Spatial registration and normalization of images.
Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate.
Gill TM, Sarter M, Givens B. Sustained visual attention performance-associated prefrontal neuronal activity: evidence for cholinergic modulation.
Goldberg E, Gerstman LJ, Mattis S, Hughes JE, Bilder RM, Jr., Sirio CA. Effects of cholinergic treatment on posttraumatic anterograde amnesia.
Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS. A voxel-based morphometric study of ageing in 465 normal adult human brains.
Gorman LK, Fu K, Hovda DA, Murray M, Traystman RJ. Effects of traumatic brain injury on the cholinergic system in the rat.
Graham DI, Gennarelli TA, McIntosh TK. Trauma. In: Graham DI and Lantos PL, editors. Greenfield's neuropathology. Vol. 1. London: Arnold;
Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, et al. The association between head trauma and Alzheimer's disease.
Greenspan L, McLellan BA, Greig H. Abbreviated injury scale and injury severity score: a scoring chart.
Hanaway J, Woolsey TA, Gado MH, Roberts MP. The brain atlas. Bethesda (MA): Fitzgerald Science Press;
Harmer CJ, McTavish SF, Clark L, Goodwin GM, Cowen PJ. Tyrosine depletion attenuates dopamine function in healthy volunteers.
Hasselmo ME. Neuromodulation: acetylcholine and memory consolidation.
Hellawell DJ, Taylor RT, Pentland B. Cognitive and psychosocial outcome following moderate or severe traumatic brain injury.
Heyman A, Wilkinson WE, Stafford JA, Helms MJ, Sigmon AH, Weinberg T. Alzheimer's disease: a study of epidemiological aspects.
Himmelheber AM, Sarter M, Bruno JP. Increases in cortical acetylcholine release during sustained attention performance in rats.
Hutchinson PJ, O'Connell MT, Al-Rawi PG, Kett-White CR, Gupta AK, Maskell LB, et al. Increases in GABA concentrations during cerebral ischaemia: a microdialysis study of extracellular amino acids.
Jakala P, Riekkinen M, Sirvio J, Koivisto E, Kejonen K, Vanhanen M, et al. Guanfacine, but not clonidine, improves planning and working memory performance in humans.
Jakala P, Sirvio J, Riekkinen M, Koivisto E, Kejonen K, Vanhanen M, et al. Guanfacine and clonidine, alpha 2-agonists, improve paired associates learning, but not delayed matching to sample, in humans.
Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury.
Jiang JY, Lyeth BG, Delahunty TM, Phillips LL, Hamm RJ. Muscarinic cholinergic receptor binding in rat brain at 15 days following traumatic brain injury.
Kemp PM, Holmes C, Hoffmann S, Wilkinson S, Zivanovic M, Thom J, et al. A randomized placebo controlled study to assess the effects of cholinergic treatment on muscarinic receptors in Alzheimer's disease.
Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system.
Kopelman MD. The cholinergic neurotransmitter system in human memory and dementia: a review.
Leonard JR, Maris DO, Grady MS. Fluid percussion injury causes loss of forebrain choline acetyltransferase and nerve growth factor receptor immunoreactive cells in the rat.
Leonard JR, Grady MS, Lee ME, Paz JC, Westrum LE. Fluid percussion injury causes disruption of the septohippocampal pathway in the rat.
Levin HS, Peters BH, Kalisky Z, High WM, Jr., von Laufen A, Eisenberg HM, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients.
Levin ED, Sledge D, Baruah A, Addy NA. Ventral hippocampal NMDA blockade and nicotinic effects on memory function.
Lucas-Meunier E, Fossier P, Baux G, Amar M. Cholinergic modulation of the cortical neuronal network.
Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review.
Lyeth BG, Jiang JY, Delahunty TM, Phillips LL, Hamm RJ. Muscarinic cholinergic receptor binding in rat brain following traumatic brain injury.
Marshall LF, Bowers Marshall S, Klauber MR, Van Berkum Clark M, Eisenberg HM, Jane JA, et al. A new classification of head injury based on computerized tomography.
Mayou R, Bryant B. Outcome in consecutive emergency department attenders following a road traffic accident.
Middleton HC, Sharma A, Agouzoul D, Sahakian BJ, Robbins TW. Idazoxan potentiates rather than antagonizes some of the cognitive effects of clonidine.
Millar K, Nicoll JA, Thornhill S, Murray GD, Teasdale GM. Long term neuropsychological outcome after head injury: relation to APOE genotype.
Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer's disease.
Muir JL, Everitt BJ, Robbins TW. AMPA-induced excitotoxic lesions of the basal forebrain: a significant role for the cortical cholinergic system in attentional function.
Muir JL, Robbins TW, Everitt BJ. Disruptive effects of muscimol infused into the basal forebrain on conditional discrimination and visual attention: differential interactions with cholinergic.
Müller U, Murai T, Bauer-Wittmund T, von Cramon DY. Paroxetine versus citalopram treatment of pathological crying after brain injury.
Murdoch I, Perry EK, Court JA, Graham DI, Dewar D. Cortical cholinergic dysfunction after human head injury.
Murdoch I, Nicoll JA, Graham DI, Dewar D. Nucleus basalis of Meynert pathology in the human brain after fatal head injury.
Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury.
Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, et al. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study.
Ono J, Yamaura A, Kubota M, Okimura Y, Isobe K. Outcome prediction in severe head injury: analyses of clinical prognostic factors.
Owen AM, Downes JJ, Sahakian BJ, Polkey CE, Robbins TW. Planning and spatial working memory following frontal lobe lesions in man.
Owen AM, Sahakian BJ, Semple J, Polkey CE, Robbins TW. Visuo-spatial short-term recognition memory and learning after temporal lobe excisions, frontal lobe excisions or amygdalo-hippocampectomy in man.
Park SB, Coull JT, McShane RH, Young AH, Sahakian BJ, Robbins TW, et al. Tryptophan depletion in normal volunteers produces selective impairments in learning and memory.
Patel HC, Menon DK, Tebbs S, Hawker R, Hutchinson PJ, Kirkpatrick PJ. Specialist neurocritical care and outcome from head injury.
Perry E, Walker M, Grace J, Perry R. Acetylcholine in mind: a neurotransmitter correlate of consciousness?
Pike BR, Hamm RJ. Chronic administration of a partial muscarinic M1 receptor agonist attenuates decreases in forebrain choline acetyltransferase immunoreactivity following experimental.
Polo MD, Newton P, Rogers D, Escera C, Butler S. ERPs and behavioural indices of long-term preattentive and attentive deficits after closed head injury.
Ragozzino ME, Unick KE, Gold PE. Hippocampal acetylcholine release during memory testing in rats: augmentation by glucose.
Rasmusson DX, Brandt J, Martin DB, Folstein MF. Head injury as a risk factor in Alzheimer's disease.
Ridley RM, Bowes PM, Baker HF, Crow TJ. An involvement of acetylcholine in object discrimination learning and memory in the marmoset.
Ridley RM, Samson NA, Baker HF, Johnson JA. Visuospatial learning impairment following lesion of the cholinergic projection to the hippocampus.
Ridley RM, Aitken DM, Baker HF. Learning about rules but not about reward is impaired following lesions of the cholinergic projection to the hippocampus.
Ridley RM, Pugh P, Maclean CJ, Baker HF. Severe learning impairment caused by combined immunotoxic lesion of the cholinergic projections to the cortex and hippocampus in monkeys.
Robertson IH, Manly T, Andrade J, Baddeley BT, Yiend J. ‘Oops!’: performance correlates of everyday attentional failures in traumatic brain injured and normal subjects.
Rogers JL, Kesner RP. Cholinergic modulation of the hippocampus during encoding and retrieval.
Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer's disease. Donepezil Study Group.
Ruff RM, Crouch JA, Troster AI, Marshall LF, Buchsbaum MS, Lottenberg S, et al. Selected cases of poor outcome following a minor brain trauma: comparing neuropsychological and positron emission tomography assessment.
Rusted J, Graupner L, Warburton D. Effects of post-trial administration of nicotine on human memory: evaluating the conditions for improving memory.
Sahakian BJ. Cholinergic drugs and human cognitive performance. In: Iversen LL, Iversen SD and Snyder SH, editors. Handbook of psychopharmacology. Vol. 20. New York: Plenum Press;
Sahakian BJ, Morris RG, Evenden JL, Heald A, Levy R, Philpot M, et al. A comparative study of visuospatial memory and learning in Alzheimer-type dementia and Parkinson's disease.
Sahakian B, Jones G, Levy R, Gray J, Warburton D. The effects of nicotine on attention, information processing, and short-term memory in patients with dementia of the Alzheimer type.
Sandel ME, Olive DA, Rader MA. Chlorpromazine-induced psychosis after brain injury.
Schmidt RH, Grady MS. Loss of forebrain cholinergic neurons following fluid-percussion injury: implications for cognitive impairment in closed head injury.
Schmidt RH, Scholten KJ, Maughan PH. Time course for recovery of water maze performance and central cholinergic innervation after fluid percussion injury.
Selden NR, Gitelman DR, Salamon-Murayama N, Parrish TB, Mesulam MM. Trajectories of cholinergic pathways within the cerebral hemispheres of the human brain.
Shalat SL, Seltzer B, Pidcock C, Baker EL Jr. Risk factors for Alzheimer's disease: a case-control study.
Signorini DF, Andrews PJ, Jones PA, Wardlaw JM, Miller JD. Predicting survival using simple clinical variables: a case study in traumatic brain injury.
Sihver S, Marklund N, Hillered L, Långström B, Watanabe Y, Bergström M. Changes in mACh, NMDA and GABA(A) receptor binding after lateral fluid-percussion injury: in vitro autoradiography of rat brain frozen sections.
Sinson G, Perri BR, Trojanowski JQ, Flamm ES, McIntosh TK. Improvement of cognitive deficits and decreased cholinergic neuronal cell loss and apoptotic cell death following neurotrophin infusion after experimental traumatic.
Smith A, Sturgess W, Rich N, Brice C, Collison C, Bailey J, et al. The effects of idazoxan on reaction times, eye movements and the mood of healthy volunteers and patients with upper respiratory tract illnesses.
Stanislav SW. Cognitive effects of antipsychotic agents in persons with traumatic brain injury.
Swainson R, Hodges JR, Galton CJ, Semple J, Michael A, Dunn BD, et al. Early detection and differential diagnosis of Alzheimer's disease and depression with neuropsychological tasks.
Sweeney JA, Kmiec JA, Kupfer DJ. Neuropsychologic impairments in bipolar and unipolar mood disorders on the CANTAB neurocognitive battery.
Takagi N, Miyake K, Taguchi T, Tamada H, Takagi K, Sugita N, et al. Failure in learning task and loss of cortical cholingergic fibers in microsphere-embolized rats.
Tavares JV, Drevets WC, Sahakian BJ. Cognition in mania and depression.
van der Naalt J, Hew JM, van Zomeren AH, Sluiter WJ, Minderhoud JM. Computed tomography and magnetic resonance imaging in mild to moderate head injury: early and late imaging related to outcome.
Vesey R, Birrell JM, Bolton C, Chipperfield RS, Blackwell AD, Dening TR, et al. Cholinergic nicotinic systems in Alzheimer's disease: prospects for pharmacological intervention.
Voytko ML. Cognitive functions of the basal forebrain cholinergic system in monkeys: memory or attention?
Voytko ML, Olton DS, Richardson RT, Gorman LK, Tobin JR, Price DL. Basal forebrain lesions in monkeys disrupt attention but not learning and memory.
Weinberg RM, Auerbach SH, Moore S. Pharmacologic treatment of cognitive deficits: a case study.
Weiss F, Porrino LJ. Behavioral neurobiology of alcohol addiction: recent advances and challenges.
Whyte J, Polansky M, Fleming M, Coslett HB, Cavallucci C. Sustained arousal and attention after traumatic brain injury.
Wurtman RJ. Choline metabolism as a basis for the selective vulnerability of cholinergic neurons.
Author notes
1Wolfson Brain Imaging Centre and Departments of 2Anaesthetics, 3Neurosurgery and 4Psychiatry, University of Cambridge, Addenbrooke's Hospital, Cambridge, UK


{kind=link}
{kind=link}