




Abstract
We assessed the impact of amyloid precursor protein (APP) gene locus duplications in early onset Alzheimer's disease in a Dutch population-based sample. Using real-time PCR and an in-house-developed multiplex amplicon quantification assay, we identified a genomic APP duplication in 1 out of 10 multigenerational families segregating early onset Alzheimer's disease. In this family, cerebral amyloid angiopathy (CAA) coincided with this disease. The duplicated genomic region included no other genes than APP and extended maximally over 0.7 Mb. In a sample of 65 familial early onset patients, we observed the same APP genomic duplication in one patient (1.7%), while in 36 isolated patients duplications in the APP locus were absent. This indicated that APP locus duplications explained <2% of familial, non-autosomal dominant Alzheimer's disease and are an infrequent cause of de novo mutation. Our findings corroborated a recent French study, and indicated that investigating genomic duplications in the APP locus in families segregating Alzheimer's disease and CAA should be considered.
Abbreviations
- APP
amyloid precursor protein
- CAA
cerebral amyloid angiopathy
- DQs
dosage quotients
- FISH
fluorescence in situ hybridization
- MAQ
multiplex amplicon quantification
- PCR
polymerase chain reaction
Introduction
Evidence is accumulating that an increase in genetic expression of the amyloid precursor protein (APP) gene, encoding the amyloid β precursor protein [MIM 104760], can cause dementia of the Alzheimer type [MIM 104300] through increased production of its pathogenic 42 amino acids proteolysis product, amyloid β42 (Aβ42). Not only missense mutations at or near the α-, β- and γ-secretase cleavage sites of APP and in the presenilins (Dermaut et al., 2005) but also increased expression of APP per se can cause an increase in concentration of Aβ42 and subsequent Aβ deposition. It has long been recognized that triplication of APP in patients with Down's syndrome leads to Alzheimer's dementia symptoms early in life through over-expression of APP (Rumble et al., 1989), followed by deposition of Aβ and neurodegeneration (Wisniewski et al., 1985). In a recent study, genomic duplications in the APP locus were reported in families segregating early onset Alzheimer's dementia with concurrent cerebral amyloid angiopathy (CAA) (Rovelet-Lecrux et al., 2006), suggesting that increased expression of APP can give rise to Alzheimer dementia pathology in the absence of a full trisomy 21. Along this line, previous studies have shown higher levels of APP mRNA in brains of patients with Alzheimer-type dementia (Cohen et al., 1988; Higgins et al., 1988; Lewis et al., 1988; Clark and Parhad, 1989; Vitek, 1989; Theuns and Van Broeckhoven, 2000), also suggesting that genetic variation in APP transcription might play a role in the pathomechanism of the disease. Indeed, in patients with early onset Alzheimer's dementia, we recently detected three mutations in the APP proximal promoter that caused a neuron-specific, nearly 2-fold increase in APP transcriptional activity in vitro, mimicking over-expression of APP as observed in trisomy 21 (Theuns et al., 2006). With the evidence that an increase in APP expression through APP genomic duplication or mutations in the APP promoter leads to early onset Alzheimer's disease, the hypothesis that Aβ plays a pivotal role in the disease aetiology gained strength. To assess the impact of APP locus duplications in early onset Alzheimer's disease, we performed a study in a Dutch population-based sample of patients with early onset Alzheimer-type dementia.
Material and methods
Patients
Patients were derived from an epidemiological study aiming to ascertain all patients with early onset Alzheimer's disease in Rotterdam and the four northern provinces of The Netherlands (Hofman et al., 1989; van Duijn et al., 1994). From 1980 to 1989 and between 1997 and 2000 all patients with a clinical diagnosis of early onset dementia were ascertained through nursing home physicians, neurologists, psychiatric institutions and social/geriatric services, and subsequently assessed for Alzheimer's disease according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association criteria (McKhann et al., 1984). Detailed medical records were available for all probands, and for affected relatives in case of an autosomal dominant pattern of inheritance. For genetic studies, 111 patients were available with a mean onset age of 56.1 years (range = 33–65), and a positive family history (i.e. at least one first-degree relative affected with either early or late-onset dementia) in 75 patients, of which 10 were probands of multigenerational families with an autosomal dominant inheritance. The criteria for autosomal dominant inheritance were (i) at least three patients with clinically diagnosed Alzheimer's disease in two or more generations and (ii) detailed medical records available on the clinical diagnosis of Alzheimer's disease in at least two affected relatives. In two families, early onset Alzheimer's disease was caused by missense mutations in presenilin 1 (PSEN1 Ala79Val, PSEN1 Tyr115Cys) (Cruts et al., 1998); one family showed significant linkage at 17q21 (Rademakers et al., 2002) and one family showed significant linkage at 7q36 (Rademakers et al., 2005). Putative causal mutations in PSEN1 were further identified in three patients, and mutations in PSEN2 in two patients in the remaining study population (Cruts et al., 1998). Missense mutations in APP were previously excluded (van Duijn et al., 1991, 1994). This study was approved by the medical ethical committee of the University of Antwerp.
Real-time polymerase chain reaction (PCR) allele quantification
Genomic DNA (gDNA) was extracted from peripheral lymphocytes. APP alleles were quantified using a SYBR® Green real-time PCR assay on the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Primers for exons 5, 11 and 18 and the promoter region of APP, for GABPA and ATP5J and for ubiquitin C (hUBC) and β2-microglobulin (hB2M) (one amplicon each) were designed with PrimerExpress software (Applied Biosystems). Primer sequences are available upon request. Human APP alleles were normalized for the housekeeping genes hUBC and hB2M. Twenty nanograms of gDNA were amplified in a 30 μl reaction containing 1× qPCR Mastermix Plus for SYBR® Green I (without UNG) (Eurogentec, Seraing, Belgium) and 400 nM of the respective forward and reverse primers using the universal amplification protocol (Applied Biosystems). Duplicate samples were quantified for patients and six cognitively healthy age-matched individuals to calculate dosage quotients (DQs). Samples of two patients with full trisomy 21 were included as positive controls.
Fluorescence in situ hybridization (FISH)
To confirm the presence of a genomic duplication of APP, FISH of both interphase nuclei and mechanically stretched metaphase chromosomes was performed, the latter permitting detection of duplications at an increased resolution (>100 kb) (Haaf and Ward, 1994; Laan et al., 1995). Mitotically active Epstein–Barr virus-transformed lymphoblasts of patients were arrested in metaphase by treatment with a 0.1 μg/ml colcemid solution and incubated in a hypotonic solution consisting of 2 mM HEPES (pH 7.3), 30 mM glycerol, 1 mM CaCl2, 0.8 mM MgCl2 and 1 M NaOH for 25 min at 37°C. The hypotonic cell suspension was diluted to 106 cells/ml. Mechanical stretching of the chromosomes was achieved by cyto-centrifuging (Cytospin 4, Shandon) to ethanol-cleaned silanized glass slides at 800–1600 r.p.m. for 5–10 min. Preparations were fixed in −20°C methanol for 30 min. In addition to stretched chromosomes, slides also contained interphase nuclei.
BAC clones RP11-15D13 and RP11-910G8, hybridizing to the 3′ and 5′ region of APP, respectively, and one reference probe (RP11-451M12) hybridizing to 21q22.12, at ∼8 Mb from APP [FISH cytogenetic reference map (Cheung et al., 2001)] were cultured, and DNA was prepared in accordance with standard procedures. Probes were labelled with digoxigenin-11-dUTP (RP11-451M12) or biotin-11-dUTP (RP11-15D13 and RP11-910G8) by nick translation. FISH was performed according to a standard protocol (Lichter et al., 1988), testing either the 3′ end APP probe or the 5′ end APP probe together with the reference probe. The biotinylated probe (RP11-910G8 or RP11-15D13) was detected using Texas Red-conjugated avidin followed by goat anti-avidin and a second layer of avidin–Texas Red. For the digoxigenin-labelled probe (RP11-451M12), mouse anti-digoxigenin followed by sheep anti-mouse-digoxigenin and FITC-conjugated sheep anti-digoxigenin was used. Chromosomal DNA was counterstained with DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride). Hybridization signals were imaged using a Zeiss Axioskop 50 fluorescent microscope (Carl Zeiss NV, Zaventem, Brussels) equipped with specific filter sets connected to a UNIX workstation with an imaging analysis program (Applied Imaging System, San Jose, CA, USA). Two patients with full trisomy 21 were included as positive control.
Multiplex amplicon quantification
To detect and delineate sizes of APP locus duplications, an in-house-developed technique for multiplex amplicon quantification (MAQ) was used (D. Goossens, in preparation), consisting of a multiplex PCR amplification of several fluorescently labelled target and reference amplicons, followed by fragment analysis on an ABI 3730 DNA analyser (Applied Biosystems). The comparison of normalized peak areas between a patient and control individuals results in a DQ of the target amplicon. Five fluorescently labelled amplicons in APP and 17 labelled amplicons in 11 surrounding genes (FLJ42200, c21orf42, MRPL39, JAM2, CYYR1, ADAMTS1, ADAMTS5, USP16, CCT8, BACH1 and GRIK1) were simultaneously amplified with 15 reference fragments randomly located on different chromosomes, using 50 ng gDNA and optimized reaction conditions (available upon request). DQs were calculated using an in-house-developed MAQ software (MAQs) package (Author Webpage). A DQ between 1.3 and 1.75 was considered indicative of a heterozygous duplication.
Results
Autosomal dominant early onset Alzheimer's disease
First, we analysed the contribution of APP locus duplications to autosomal dominant early onset Alzheimer's disease in the Dutch population-based sample. Real-time PCR quantification of APP in 10 probands revealed a DQ > 1.5 for all three APP exons 5, 11 and 18 in the proband of one family, 1104, pointing to a heterozygous duplication of APP.
Family 1104 is a four-generation pedigree including 10 relatives (77.8% female) with dementia, and a mean onset age of 52.1 years [standard deviation (SD) = 7.9; range = 40–62] and mean disease duration of 8.2 years (SD = 4.7) (Fig. 1). Clinical characteristics of Family 1104 are summarized in Table 1. The proband, III.14, first complained of memory impairment at 53 years, interfering with daily activities. The symptoms were slowly progressive, and affecting other areas of cognition, resulting in short- and long-term memory loss, disorientation, dysphasia, apraxia and roaming behaviour. At 62 years she was admitted to a nursing home, where recurrent convulsions were observed. She died of dehydration at 66 years. Two siblings, III.15 and III.20, and their mother, II.6, had Alzheimer's disease as well. Patient III.20 had an earlier onset of symptoms, at 47 years, possibly accelerated by alcohol abuse. He had marked deficits of memory and orientation, aphasia and apraxia, agnosia of the body, involuntary movements of the chest and limbs and convulsions. He died at 51 years of a status epilepticus. Patient III.15 had an onset age of dementia of 62 years and died 2 years later. His symptoms were compatible with early onset Alzheimer's disease and included memory impairment, deficits in orientation and delusions. Of interest, two siblings, III.17 and III.19, without symptoms of dementia suffered (unspecified) strokes at an early age (35 and 56 years, respectively). None of the patients with Alzheimer's disease had a history of stroke or intracerebral haemorrhage. One sibling died of epilepsy at 24 years and one sibling was cognitively healthy until 58 years, but was lost to follow-up. The proband's grandfather, three maternal aunts and two cousins had early onset dementia as well. Patient III.13 had autopsy-confirmed Lewy body-variant Alzheimer's disease, clinically characterized by dementia with hallucinations and rigidity. Braak stage was VI C. Lewy bodies were present in the brainstem and limbic area. Brain autopsies were also previously performed in the proband's mother, II.6, and Patient III.20. Neuropathological examination of Patient III.20 demonstrated severe cortical neuronal loss, neurofibrillary tangles and abundant amyloid depositions in neuritic plaques as well as in arteries and leptomeninges, and Braak stage compatible with VI C. The inferior temporal area was most severely affected, but frontal and temporal areas, motor cortex, hippocampus and corpus striatum showed widespread pathology as well. Loss of ganglion cells in the hippocampus, though pronounced, was relatively moderate when compared with other areas. CAA was present in all areas, but was most abundant in striate and parastriate areas (Brodmann area = 17, 18). No Lewy bodies were observed. In a report of the neuropathological examination of the proband's mother, II.6, in 1969, severe atrophy of the brain (weight = 880 g) and abundant amyloid depositions in cortical areas and in vessel walls were mentioned.
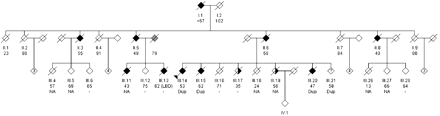
Pedigree of Family 1104. Black filled symbols indicate patients, half-filled symbols indicate individuals with a history of stroke, grey filled symbol indicates those reported to be forgetful, but not fulfilling criteria for dementia. Numbering is in accordance with previous publication of this pedigree, except III.17, III.18 and III.19 (previously III.18, III.19 and III.17) (van Duijn et al., 1994). The proband is indicated with an arrowhead. Age at onset is given for relatives with dementia or stroke; age at death or last examination is given for unaffected relatives. Presence or absence of APP genomic duplication is given for the third and fourth generation. ‘Dup’ indicates heterozygous duplication, ‘-’ indicates no duplication, ‘NA’ indicates no DNA available.
Clinical characteristics of patients in Family 1104
| Patient | Gender | Age (years) at | Disease duration | APOE genotype* | Presenting symptoms | Structural neuroimaging | Autopsy performed | Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| onset | death | ||||||||
| I.1 | M | ? | 67 | ? | Dementia** | ||||
| II.3 | F | 55 | 66 | 11 | Dementia | ||||
| II.5 | F | 49 | 54 | 5 | Dementia | ||||
| II.6 | F | 58 | 65 | 7 | Impaired memory | Yes | Definite AD + CAA | ||
| II.8 | F | 40 | 57 | 17 | Dementia | ||||
| III.11 | F | 43 | 50 | 7 | Dementia | ||||
| III.13 | F | 62 | 70 | 8 | Impaired memory, hallucinations | Yes | LBD | ||
| III.14 | F | 53 | 66 | 13 | 33 | Impaired memory and imprinting | Global cortical and subcortical atrophy (CT) | Probable AD | |
| III.15 | M | 62 | 64 | 2 | 33 | Impaired memory and imprinting, disorientation | Probable AD | ||
| III.20 | M | 47 | 51 | 4 | 33 | Impaired memory and imprinting, paranoia | Global cortical and subcortical atrophy, right > left (CT) | Yes | Definite AD + CAA |
| Patient | Gender | Age (years) at | Disease duration | APOE genotype* | Presenting symptoms | Structural neuroimaging | Autopsy performed | Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| onset | death | ||||||||
| I.1 | M | ? | 67 | ? | Dementia** | ||||
| II.3 | F | 55 | 66 | 11 | Dementia | ||||
| II.5 | F | 49 | 54 | 5 | Dementia | ||||
| II.6 | F | 58 | 65 | 7 | Impaired memory | Yes | Definite AD + CAA | ||
| II.8 | F | 40 | 57 | 17 | Dementia | ||||
| III.11 | F | 43 | 50 | 7 | Dementia | ||||
| III.13 | F | 62 | 70 | 8 | Impaired memory, hallucinations | Yes | LBD | ||
| III.14 | F | 53 | 66 | 13 | 33 | Impaired memory and imprinting | Global cortical and subcortical atrophy (CT) | Probable AD | |
| III.15 | M | 62 | 64 | 2 | 33 | Impaired memory and imprinting, disorientation | Probable AD | ||
| III.20 | M | 47 | 51 | 4 | 33 | Impaired memory and imprinting, paranoia | Global cortical and subcortical atrophy, right > left (CT) | Yes | Definite AD + CAA |
*APOE genotypes were determined previously (van Duijn et al., 1994); **dementia = diagnosis obtained through family informant; AD = Alzheimer's disease; CAA = cerebral amyloid angiopathy; LBD = Lewy body-variant Alzheimer's disease.
Clinical characteristics of patients in Family 1104
| Patient | Gender | Age (years) at | Disease duration | APOE genotype* | Presenting symptoms | Structural neuroimaging | Autopsy performed | Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| onset | death | ||||||||
| I.1 | M | ? | 67 | ? | Dementia** | ||||
| II.3 | F | 55 | 66 | 11 | Dementia | ||||
| II.5 | F | 49 | 54 | 5 | Dementia | ||||
| II.6 | F | 58 | 65 | 7 | Impaired memory | Yes | Definite AD + CAA | ||
| II.8 | F | 40 | 57 | 17 | Dementia | ||||
| III.11 | F | 43 | 50 | 7 | Dementia | ||||
| III.13 | F | 62 | 70 | 8 | Impaired memory, hallucinations | Yes | LBD | ||
| III.14 | F | 53 | 66 | 13 | 33 | Impaired memory and imprinting | Global cortical and subcortical atrophy (CT) | Probable AD | |
| III.15 | M | 62 | 64 | 2 | 33 | Impaired memory and imprinting, disorientation | Probable AD | ||
| III.20 | M | 47 | 51 | 4 | 33 | Impaired memory and imprinting, paranoia | Global cortical and subcortical atrophy, right > left (CT) | Yes | Definite AD + CAA |
| Patient | Gender | Age (years) at | Disease duration | APOE genotype* | Presenting symptoms | Structural neuroimaging | Autopsy performed | Diagnosis | |
|---|---|---|---|---|---|---|---|---|---|
| onset | death | ||||||||
| I.1 | M | ? | 67 | ? | Dementia** | ||||
| II.3 | F | 55 | 66 | 11 | Dementia | ||||
| II.5 | F | 49 | 54 | 5 | Dementia | ||||
| II.6 | F | 58 | 65 | 7 | Impaired memory | Yes | Definite AD + CAA | ||
| II.8 | F | 40 | 57 | 17 | Dementia | ||||
| III.11 | F | 43 | 50 | 7 | Dementia | ||||
| III.13 | F | 62 | 70 | 8 | Impaired memory, hallucinations | Yes | LBD | ||
| III.14 | F | 53 | 66 | 13 | 33 | Impaired memory and imprinting | Global cortical and subcortical atrophy (CT) | Probable AD | |
| III.15 | M | 62 | 64 | 2 | 33 | Impaired memory and imprinting, disorientation | Probable AD | ||
| III.20 | M | 47 | 51 | 4 | 33 | Impaired memory and imprinting, paranoia | Global cortical and subcortical atrophy, right > left (CT) | Yes | Definite AD + CAA |
*APOE genotypes were determined previously (van Duijn et al., 1994); **dementia = diagnosis obtained through family informant; AD = Alzheimer's disease; CAA = cerebral amyloid angiopathy; LBD = Lewy body-variant Alzheimer's disease.
Segregation and delineation of the APP locus duplication in Family 1104
For segregation analysis, DNA of the proband and two affected relatives and six at-risk individuals in generations III and IV was available. In these, both real-time PCR quantification of APP and MAQ PCR of APP and 11 surrounding genes showed an increased DQ for all APP amplicons in the two affected relatives (III.15 and III.20; mean DQ over all MAQ APP amplicons was 1.6 and 1.5, respectively), but for none of the surrounding genes. Also, one unaffected sibling (III.21; mean DQ = 1.6) aged 58 years, and still within the ±1 SD risk age range of the disease, carried the APP duplication. Two unaffected siblings and three unaffected cousins aged >64 years were negative (mean DQ ranging from 0.9 to 1.0). Together, these data suggested that the APP duplication segregated with the disease. Of note, the APP duplication was not present in Sibling III.17 who had a history of (unspecified) stroke at an early age.
Standard metaphase FISH of lymphoblasts of the proband and her two affected siblings did not reveal the APP duplication owing to limited resolution, but in interphase nuclei duplicated signals resulting from the APP containing probe (for both the 5′ end and the 3′ end probe) could consistently be observed (Fig. 2A and B). After cyto-centrifuging in order to mechanically stretch the chromosomes, which allows the detection of smaller duplicated regions, the presence of the APP duplication could be confirmed (both with the 5′ end and the 3′ end probe) in the proband and both affected siblings, indicative of a tandem duplication of APP on chromosome 21 (Fig. 2C).
![FISH analysis of an interphase nucleus (A and B) and a mechanically stretched metaphase chromosome of an APP genomic duplication patient of Family 1104 (C). Red signals indicate APP [BAC clones RP11-15D13 (A and C) or RP11-910G8 (B)] and the green signal indicates a 21q22.12 reference probe (RP11-451M12). (Interphase nuclei showed a slight degree of mechanical stretching due to cyto-centrifuging.)](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/129/11/10.1093/brain/awl203/2/m_awl203f2.gif?Expires=1716312844&Signature=H6N5dtXLnbo7ZroV911UZzuIBaOd0HlmnzQpcAbpCyxRRuYqDl9Hifj3x6Hl7n8A9KDursiVfat8rRmogQ4EwCOaWX7n~PFE8vq0olE0fbE2Yj23UOW36l06bnc-M0jtdW90VtLM4t0smbAmAxKFMBfpjM-96jfLB0GKSdpFVjYUVUOP~zyXT-K-~3XhqYAtcL1Kh7K8P~tKV-MQSVoTkZX8hHOu1973zNseaopDD0b2~Mbw-uLtPtXnkaHRtMzHhXO3OwAvquRFv5pRmsLE6rVBR48lgTVQhUjiCPZTWRVDBTC321pTTpuu3urYIXCkvA~3H~pQavfoNyRGpbEalA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FISH analysis of an interphase nucleus (A and B) and a mechanically stretched metaphase chromosome of an APP genomic duplication patient of Family 1104 (C). Red signals indicate APP [BAC clones RP11-15D13 (A and C) or RP11-910G8 (B)] and the green signal indicates a 21q22.12 reference probe (RP11-451M12). (Interphase nuclei showed a slight degree of mechanical stretching due to cyto-centrifuging.)
On the basis of the MAQ PCR assay of APP and 11 surrounding genes covering a region of 5.51 Mb, we delineated the size of the genomic duplication to a fragment of 290–750 kb, including APP (Fig. 3). Real-time PCR of the APP promoter, and GABPA and ATP5J, two genes for which no amplicons were included in the MAQ PCR assay, confirmed that the genomic duplication included APP and its promoter region, but no other genes.
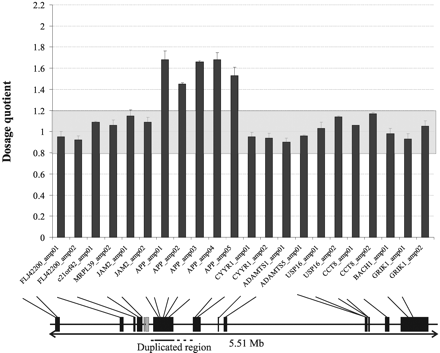
(Upper panel): Dosage plot for the Proband III.14 of Family 1104. DQs are presented with their standard error. DQs for each amplicon were calculated using 4 control individuals and 15 random control amplicons. Only amplicons tested with MAQ are shown. The grey area in the plot (DQ = 0.8–1.2) represents normal variation. DQs between 1.3 and 1.75 are indicative of a heterozygous duplication. (Lower panel): Genes analysed. Black boxes represent genes analysed by MAQ and grey boxes represent genes analysed by SYBR Green real-time PCR (GABPA, ATP5J). Solid horizontal bar = minimal duplicated region, dotted lines = interval of duplication boundaries.
Population-based early onset Alzheimer's disease
Using the MAQ assay, we analysed the remaining 101 patients from the same geographical area as Family 1104, including 65 patients with at least one first-degree relative with Alzheimer's disease. We observed evidence of a genomic duplication in one familial patient [1043; mean DQ over all MAQ APP amplicons = 1.35 (SE = 0.05)]. Similar to the duplication in Family 1104, the duplication in Patient 1043 included APP but none of the adjacent genes. The presence of the APP duplication was confirmed by SYBR Green real-time PCR. Analysis of amplicons located in the APP promoter, and in GABPA and ATP5J, was positive only for the APP promoter, indicating a maximally duplicated region of 0.7 Mb.
Patient 1043 presented at 57 years with pronounced expressive and receptive dysphasia, difficulty finding words and naming objects and forgetfulness existing for 2 years. In a period of 3 years he became fully ADL-dependent with distinct apraxia and agnosia of the body, and impairment of both short-term and long-term memory. Later in the course of the disease he developed severe rigidity of all limbs, convulsions and myoclonia. A CT scan revealed extensive atrophy without focal lesions. He carried the APOE ɛ3ɛ4 genotype. Family history was positive in the second degree. His father died of old age, but his mother died at 63 years. An affected maternal uncle (age at onset = 59 years) had a stroke during the course of the disease. Patient 1043 died without autopsy, at 67 years, of pneumonia.
Discussion
In this Dutch population-based sample of early onset dementia we identified an APP duplication in a family with a segregation pattern compatible with autosomal dominant inheritance, and neuropathology compatible with Alzheimer's disease with extensive CAA. Our findings are in agreement with the recent observation of APP locus duplications in French families with autosomal dominant early onset dementia of the Alzheimer type and concurrent CAA (Rovelet-Lecrux et al., 2006). In this study a genomic duplication in the APP locus was present in 5 out of 65 families (∼8%) with autosomal dominant early onset Alzheimer's disease. We detected the APP duplication in 1 out of 10 (10%) of the autosomal dominant early onset Alzheimer's disease families in our sample. Although numbers are small, together these data indicated that investigation of genomic duplications in the APP locus in multiplex early onset families is warranted when simple mutations in known Alzheimer's disease genes have been excluded. Fast and reliable screening for these genomic duplications is now feasible owing to the development of our MAQ assay, which not only allows detection of a genomic duplication but also provides information of the size of the duplication and genes involved. In 65 patients with a positive family history, we detected one other similar sized genomic duplication. In this Dutch population-based sample, genomic duplications occurred at a frequency of 2/111 (1.8%) overall, and at a frequency of 2.7% (2/75) in familial Alzheimer's disease patients. With a frequency of 10% in multiplex early onset families, the frequency increases with increasing evidence of a genetic background. In 36 patients with sporadic early onset Alzheimer's disease, we did not detect duplications, suggesting that de novo genomic APP duplications are unlikely to be a frequent cause of early onset Alzheimer-type dementia.
In contrast to the French families, in which the duplication extended over larger genomic regions ranging from minimally 0.58 to 6.37 Mb and included several other genes apart from APP (Rovelet-Lecrux et al., 2006), the duplication in Dutch Family 1104, and Patient 1043, contained only APP and extended over maximally 0.7 Mb. Thus, our data showed that a genomic duplication of APP is sufficient to cause the mixed phenotype of Alzheimer's disease and CAA without contribution from any of the adjacent genes. Further, the difference in duplication sizes between carriers indicated that there is no single genomic architectural feature in the APP locus serving as a recombination substrate, but rather that APP is located in a ‘hotspot’ region of increased recombination, owing to the presence of elements like low copy repeats. Further, genomic fine mapping will be needed to clarify the mechanism of genomic duplications in the APP locus. The fact that we observed a genomic duplication limited to APP in two seemingly unrelated early onset patients might be explained by the presence of an unknown common founder. The geographic region from which the two patients originated is characterized by limited immigration, and we have already previously observed a founder effect in patients from this region carrying a PSEN1 Ala79Val mutation or Glu318Gly substitution (Cruts et al., 1998; Dermaut et al., 1999).
Earlier, it has been suggested that trisomy 21 mosaicism in brain tissue might contribute to Alzheimer's disease (Geller and Potter, 1999). With the techniques we used we have been unable to accurately study mosaicism, but in light of the recent findings that APP genomic duplication and APP promoter mutations increasing APP transcription can cause Alzheimer's disease (Rovelet-Lecrux et al., 2006; Theuns et al., 2006), this hypothesis might gain a renewed interest. Although autopsies were performed in the past in two APP duplication carriers of Family 1104, brain material was no longer available for detailed expression studies.
Of note, several of the patients here described had recurrent seizures or convulsions during the course of their disease. Possibly, seizures are part of the clinical phenotype caused by APP genomic duplication, owing to increased amyloid pathology or petechial haemorrhage following CAA. Seizures are a common symptom in Alzheimer's disease per se, and numbers in our study are too small to reach a firm conclusion; nevertheless, this observation warrants further attention in future clinical studies of APP duplication carriers.
Recent studies support the fact that small genomic rearrangements like duplication and deletions are much more frequent than once thought (Feuk et al., 2006). In other neurodegenerative proteopathies like Parkinson's disease, the role of gene dosage in the pathomechanism has longer been recognized (Eriksen et al., 2005). In the same Dutch population-based sample we previously identified three patients carrying an APP promoter mutation affecting gene expression (Theuns et al., 2006), whereas the frequency of APP missense mutations in this population was 0% (van Duijn et al., 1991, 1994). Therefore, with 5 out of 111 (4.5%) patients in this Dutch population either carrying an APP genomic duplication or APP promoter mutation, mutations affecting expression of APP are a more frequent cause of early onset Alzheimer-type dementia. This underscores the fact that mutations affecting expression, either regulatory or copy number polymorphisms, merit molecular investigations in Alzheimer's disease patients and families.
We are grateful to the participants of this study for their cooperation. We also acknowledge the Genetic Service Facility (Author Webpage) for the genetic analyses. This work was supported by the Fund for Scientific Research-Flanders (FWO-F); the Interuniversity Attraction Poles (IUAP) program P5/19 of the Belgian Federal Science Policy Office, Belgium; the EU contract LSHM-CT-2003-503330 (APOPIS); the Industrial Research Fund of the University of Antwerp (IOF) and The Netherlands Organization for Scientific Research (NWO); and the Center for Medical Systems Biology (CMSB). J.T. and M.C. are postdoctoral fellows, and N.B. and I.G. are PhD fellows of the FWO-F. Funding to pay the Open Access publication charges for this article was provided by the Special Research Fund of the University of Antwerp.


{kind=link}
{kind=link}
{kind=link}