




Abstract
The most common pathology in frontotemporal dementia (FTD) is tau-negative, ubiquitin-immunoreactive (ub-ir) neuronal inclusions (FTLD-U). Recently, we identified mutations in the progranulin (PGRN) gene as the cause of autosomal dominant FTLD-U linked to chromosome 17. Here, we describe the neuropathology in 13 patients from 6 different families, each with FTD caused by a different PGRN mutation. The most consistent feature was the presence of ub-ir lentiform neuronal intranuclear inclusions (NII) in the neocortex and striatum. In addition, the neocortex showed moderate-to-severe superficial laminar spongiosis, chronic degenerative changes, ub-ir neurites and well-defined ub-ir neuronal cytoplasmic inclusions (NCI). In the striatum, there were numerous ub-ir neurites. NCI in the hippocampus usually had a granular appearance. In contrast, familial FTLD-U cases without PGRN mutations had no NII, less severe neocortical and striatal pathology and hippocampal NCI that were more often solid. Eight cases in which genetic analysis was not available also had NII and an overall pathology similar to those with proven mutations. None of our cases of FTLD-U without NII showed the same pattern of pathology as those with mutations. These findings suggest that FTD caused by PGRN mutations has a recognizable pathology with the most characteristic feature being ub-ir NII.
Abbreviations
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- FTLD-U
FTLD with ubiquitin-positive inclusions
- MAPT
microtubule-associated protein tau
- MND
motor neuron disease
- NCI
neuronal cytoplasmic inclusions
- NII
neuronal intranuclear inclusions
- PGRN
progranulin
- ub-ir
ubiquitin-immunoreactive
Introduction
Frontotemporal dementia (FTD) is a clinical syndrome characterized by abnormalities in personality, behaviour and language, with relative preservation of episodic memory (The Lund and Manchester Groups, 1994; Neary et al., 1998; McKhann et al., 2001). Some patients with FTD also develop a pyramidal or extrapyramidal movement disorder (The Lund and Manchester Groups, 1994; McKhann et al., 2001). FTD accounts for 5–15% of dementia and is the second commonest type with presenile onset (Neary et al., 1998; Ratnavalli et al., 2002; Bird et al., 2003; Rosso et al., 2003). Several different patterns of neuropathology may be associated with clinical FTD; the common feature being degeneration of the frontal and temporal lobes [frontotemporal lobar degeneration (FTLD)] (The Lund and Manchester Groups, 1994; McKhann et al., 2001; Trojanowski and Dickson, 2001). Many cases are characterized by the abnormal accumulation of hyperphosphorylated tau protein in the cytoplasm of neurons and glial cells. However, the majority of FTD is not associated with tauopathy (Josephs et al., 2004; Lipton et al., 2004; Johnson et al., 2005). In the past, these tau-negative cases were usually classified as ‘dementia lacking distinctive histopathology’ (DLDH), because of the absence of any specific changes identified with special histochemical stains (Knopman et al., 1990). More recently, however, a growing number of cases of FTD are being recognized as having neuronal cytoplasmic inclusions (NCI) that are ubiquitin-immunoreactive (ub-ir) but negative for tau, α-synuclein or intermediate filament proteins (FTLD-U). This pattern of pathology was first recognized in patients with motor neuron disease (MND) and dementia (Okamoto et al., 1992; Wightman et al., 1992) but was subsequently found in patients with FTD in the absence of motor dysfunction (Jackson et al., 1996). Several recent studies have shown that the majority of cases previously classified as DLDH are in fact FTLD-U and that FTLD-U is the most common pathology underlying FTD (Josephs et al., 2004a; Lipton et al., 2004; Johnson et al., 2005; Mackenzie et al., 2006b).
FTD is often familial (25–50%), usually with an autosomal dominant pattern of inheritance and high penetrance (Stevens et al., 1998; Chow et al., 1999; McKhann et al., 2001; Bird et al., 2003; Rosso et al., 2003). It is genetically complex, with linkage studies revealing FTD loci and genes on chromosome 3p (Skibinski et al., 2005), chromosome 9q (Hosler et al., 2000), chromosome 9p (two loci) (Watts et al., 2004; Morita et al., 2006; Vance et al., 2006) and chromosome 17q (two loci) (Bird et al., 1997; Foster et al., 1997; Froelich et al., 1997; Hutton et al., 1998; Lendon et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998; Kertesz et al., 2000; Rosso et al., 2001; Rademakers et al., 2002; Mackenzie et al., 2006a). At a consensus conference in 1996, the autosomal dominant form of FTD associated with loci on chromosome 17q21 was given the overall term FTD with parkinsonism linked to chromosome 17 (FTDP-17) (Foster et al., 1997). In 1998, mutations in the microtubule-associated protein tau (MAPT) gene were first discovered in a proportion of these FTDP-17 families (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). To date, more than 30 different confirmed pathogenic mutations in MAPT have been identified in over 100 families world-wide (Rademakers et al., 2004). FTD patients with MAPT mutations consistently show tau-positive inclusion pathology at autopsy (Ghetti et al., 2003). However, MAPT mutations explain only 10–20% of all familial FTD (Poorkaj et al., 2001; Rademakers et al., 2004) and not all cases of familial FTD with tau pathology have defined MAPT mutations. Moreover, there are a number of FTD families reported, with significant linkage to the same chromosomal region on 17q21 as the MAPT gene (D17S1787–D17S806), but in which no MAPT mutation has been identified (Bird et al., 1997; Froelich et al.,1997; Lendon et al., 1998; Kertesz et al., 2000; Rosso et al., 2001; Rademakers et al., 2002; Mackenzie et al., 2006a). Importantly, all these families lack significant tau pathology, but instead demonstrate FTLD-U.
We recently demonstrated that mutations in the progranulin (PGRN) gene are the cause of autosomal dominant FTLD-U linked to chromosome 17 (Baker et al., 2006). PGRN is a multifunctional secreted growth factor that mediates cell cycle progression and cell motility (He and Bateman, 2003). It is expressed in a variety of tissues and is involved in development, wound repair and inflammation. The function of PGRN in the nervous system is not yet fully understood but it is normally expressed by a subset of pyramidal neurons and is upregulated in activated microglia (Daniel et al., 2000). In our previous study, the majority of PGRN mutations, identified in FTD families, resulted in mRNAs with premature stop codons that were degraded by nonsense-mediated decay (null mutations) (Baker et al., 2006). We further demonstrated that, in these families, FTD was likely resulting from a loss of functional PGRN (haploinsufficiency), rather than the accumulation of mutant protein (Baker et al., 2006).
Here, we describe the detailed neuropathology in 13 patients from 6 different families, each family with FTD caused by a different PGRN mutation. Our findings demonstrate that PGRN mutations result in a unique pattern of neuropathology that is distinguishable from other subtypes of FTLD-U.
Material and methods
We retrospectively reviewed the neuropathology of 13 patients, from 6 different families with autosomal dominant FTD, in which PGRN mutations have been identified (Table 1) (Baker et al., 2006; Gass et al., 2006). A description of the pathology in one of these families (UBC-17, Cases 1–4) has been reported previously, prior to identification of the gene (Mackenzie et al., 2006a). The mutation in each of these six families was known to be in a different region of the PGRN gene. Twelve of the cases had undergone autopsy, while the other was a neurosurgical biopsy specimen (Case 10). The pattern of pathology in cases with proven mutations was compared with 31 other autopsy cases in which a diagnosis of FTLD-U had previously been made, based on the presence of ub-ir, tau-, α-synuclein-, intermediate filament-negative neurites and NCI in the superficial layers cerebral neocortex, dentate granule cells or both (Okamoto et al., 1992; Wightman et al., 1992; Jackson et al., 1996). These other cases had a clinical history of FTD (n = 14) or MND-dementia (n = 17) and 12 had a positive family history of similar disease (4 familial FTD, 8 familial MND-dementia). In five of these familial cases, analysis of the PGRN gene had been performed and failed to identify any abnormality.
Study subjects with identified PGRN mutations
| Case | Family | Sex | Clinical presentation | Onset (years) | Death (years) | Duration (years) | PGRN mutation |
|---|---|---|---|---|---|---|---|
| 1 | UBC17 | M | FTD, parkinsonism | 65 | 69 | 4 | 90_91insCTGC |
| 2 | UBC17 | M | FTD | 60 | 66 | 6 | 90_91insCTGC |
| 3 | UBC17 | F | FTD, weakness | 53 | 60 | 7 | 90_91insCTGC |
| 4 | UBC17 | M | FTD | 57 | 61 | 4 | 90_91insCTGC |
| 5 | UBC15 | F | FTD | 50 | 58 | 8 | 1252C→T |
| 6 | UBC15 | F | Dementia, MS | 69 | 77 | 8 | 1252C→T |
| 7 | UBC15 | M | FTD | 52 | 58 | 6 | 1252C→T |
| 8 | UBC15 | M | Dementia | 66 | 73 | 7 | 1252C→T |
| 9 | UBC14 | M | PPA, FTD | 59 | 62 | 3 | 463-1G→A (IVS4-1G→A) |
| 10 | UBC14 | F | FTD | 55 | 58 | 3 | IVS4-1G→A |
| 11 | UBC19 | F | FTD | 56 | 61 | 5 | 933+1G→A (IVS8+1G→A) |
| 12 | UBC4 | F | FTD, CBS | 65 | 71 | 6 | 1157G→A |
| 13 | UBC11 | M | FTD | 55 | 63 | 8 | 388_391delCAGT |
| 7M:6F | 58 (50–69) | 64 (58–77) | 6 (3–8) |
| Case | Family | Sex | Clinical presentation | Onset (years) | Death (years) | Duration (years) | PGRN mutation |
|---|---|---|---|---|---|---|---|
| 1 | UBC17 | M | FTD, parkinsonism | 65 | 69 | 4 | 90_91insCTGC |
| 2 | UBC17 | M | FTD | 60 | 66 | 6 | 90_91insCTGC |
| 3 | UBC17 | F | FTD, weakness | 53 | 60 | 7 | 90_91insCTGC |
| 4 | UBC17 | M | FTD | 57 | 61 | 4 | 90_91insCTGC |
| 5 | UBC15 | F | FTD | 50 | 58 | 8 | 1252C→T |
| 6 | UBC15 | F | Dementia, MS | 69 | 77 | 8 | 1252C→T |
| 7 | UBC15 | M | FTD | 52 | 58 | 6 | 1252C→T |
| 8 | UBC15 | M | Dementia | 66 | 73 | 7 | 1252C→T |
| 9 | UBC14 | M | PPA, FTD | 59 | 62 | 3 | 463-1G→A (IVS4-1G→A) |
| 10 | UBC14 | F | FTD | 55 | 58 | 3 | IVS4-1G→A |
| 11 | UBC19 | F | FTD | 56 | 61 | 5 | 933+1G→A (IVS8+1G→A) |
| 12 | UBC4 | F | FTD, CBS | 65 | 71 | 6 | 1157G→A |
| 13 | UBC11 | M | FTD | 55 | 63 | 8 | 388_391delCAGT |
| 7M:6F | 58 (50–69) | 64 (58–77) | 6 (3–8) |
Study subjects with identified PGRN mutations
| Case | Family | Sex | Clinical presentation | Onset (years) | Death (years) | Duration (years) | PGRN mutation |
|---|---|---|---|---|---|---|---|
| 1 | UBC17 | M | FTD, parkinsonism | 65 | 69 | 4 | 90_91insCTGC |
| 2 | UBC17 | M | FTD | 60 | 66 | 6 | 90_91insCTGC |
| 3 | UBC17 | F | FTD, weakness | 53 | 60 | 7 | 90_91insCTGC |
| 4 | UBC17 | M | FTD | 57 | 61 | 4 | 90_91insCTGC |
| 5 | UBC15 | F | FTD | 50 | 58 | 8 | 1252C→T |
| 6 | UBC15 | F | Dementia, MS | 69 | 77 | 8 | 1252C→T |
| 7 | UBC15 | M | FTD | 52 | 58 | 6 | 1252C→T |
| 8 | UBC15 | M | Dementia | 66 | 73 | 7 | 1252C→T |
| 9 | UBC14 | M | PPA, FTD | 59 | 62 | 3 | 463-1G→A (IVS4-1G→A) |
| 10 | UBC14 | F | FTD | 55 | 58 | 3 | IVS4-1G→A |
| 11 | UBC19 | F | FTD | 56 | 61 | 5 | 933+1G→A (IVS8+1G→A) |
| 12 | UBC4 | F | FTD, CBS | 65 | 71 | 6 | 1157G→A |
| 13 | UBC11 | M | FTD | 55 | 63 | 8 | 388_391delCAGT |
| 7M:6F | 58 (50–69) | 64 (58–77) | 6 (3–8) |
| Case | Family | Sex | Clinical presentation | Onset (years) | Death (years) | Duration (years) | PGRN mutation |
|---|---|---|---|---|---|---|---|
| 1 | UBC17 | M | FTD, parkinsonism | 65 | 69 | 4 | 90_91insCTGC |
| 2 | UBC17 | M | FTD | 60 | 66 | 6 | 90_91insCTGC |
| 3 | UBC17 | F | FTD, weakness | 53 | 60 | 7 | 90_91insCTGC |
| 4 | UBC17 | M | FTD | 57 | 61 | 4 | 90_91insCTGC |
| 5 | UBC15 | F | FTD | 50 | 58 | 8 | 1252C→T |
| 6 | UBC15 | F | Dementia, MS | 69 | 77 | 8 | 1252C→T |
| 7 | UBC15 | M | FTD | 52 | 58 | 6 | 1252C→T |
| 8 | UBC15 | M | Dementia | 66 | 73 | 7 | 1252C→T |
| 9 | UBC14 | M | PPA, FTD | 59 | 62 | 3 | 463-1G→A (IVS4-1G→A) |
| 10 | UBC14 | F | FTD | 55 | 58 | 3 | IVS4-1G→A |
| 11 | UBC19 | F | FTD | 56 | 61 | 5 | 933+1G→A (IVS8+1G→A) |
| 12 | UBC4 | F | FTD, CBS | 65 | 71 | 6 | 1157G→A |
| 13 | UBC11 | M | FTD | 55 | 63 | 8 | 388_391delCAGT |
| 7M:6F | 58 (50–69) | 64 (58–77) | 6 (3–8) |
Immunohistochemistry
Immunohistochemistry was performed on 5 μm sections of formalin fixed, paraffin-embedded tissue representing frontal and temporal neocortex, hippocampus, striatum, lower brainstem and spinal cord, when available. All immunohistochemistry, with the exception of that for PGRN, was performed using the Ventana BenchMark® XT automated staining system (Ventana, Tucson, AZ). The primary antibodies employed recognized the following proteins: ubiquitin (anti-ubiquitin, DAKO, Glostrup, Denmark; 1:500, following microwave antigen retrieval), non-phosphorylated neurofilament (anti-neurofilament protein, DAKO, Glostrup, Denmark; 1:2000, following protease digestion), phosphorylated neurofilament (SMI 31, Sternberger, Lutherville, MD; 1:8000, following protease digestion), hyperphosphorylated tau (AT-8, Innogenetics, Gent, Belgium; 1:2000 following microwave antigen retrieval and TAU-2, Sigma, Oakville, Canada; 1:1000 with 3 h initial incubation at room temperature), α-synuclein (anti-α-synuclein, Zymed, San Francisco, CA; 1:10?000, following microwave antigen retrieval), amyloid-β protein (anti-beta amyloid, DAKO, Glostrup, Denmark; 1:100 with formic acid pre-treatment and initial incubation for 3 h at room temperature) and polyglutamine containing proteins (1C2, Chemicon, Temecula, CA; 1:1000, 24 h at room temperature, following formic acid pre-treatment). Microwave antigen retrieval was done in a microwave pressure cooker for 13 min in citrate buffer, pH 6.0. Sections were developed using aminoethylcarbizole (AEC) and counterstained with haematoxylin.
PGRN immunohistochemistry was performed manually on sections of frontal neocortex and hippocampus from all cases with proven PGRN mutations. A panel of antibodies was used that recognize all regions of the PGRN protein: N-terminus (acrogranin N-19; Santa Cruz Biotechnology, Santa Cruz, CA; 1:100), C-terminus (acrogranin S-15; Santa Cruz Biotechnology, Santa Cruz, CA; 1:100 and anti-PCDGF; Zymed, South San Francisco, CA; 1:100) and the entire recombinant human protein (anti-human PGRN; R & D Systems, Minneapolis, MN; 1:500). Microwave pre-treated sections (as above) were incubated with the primary antibodies for 1 h at room temperature. Sections were then incubated with 5% biotinylated universal secondary antibody (Vector Laboratories, Burlingame, CA; 10 min), followed by streptavidin/peroxidase complex working solution (5 min) and then developed using diaminobenzidine (5 min). All sections were then counterstained with haematoxylin.
Ub-ir pathological changes were graded using the following semiquantitative system: −, none; +, rare (pathological lesions were only found in a small proportion of 20× microscopic fields examined); ++, mild (a small number of pathological structures were present in most 20× fields); +++, moderate (moderate numbers of pathological structures were present in virtually every 20× field examined); and ++++, severe (large numbers of pathological structures were present in virtually every field 20× examined). The reliability and reproducibility of this grading system has been demonstrated previously (Mackenzie and Feldman, 2004). The final score was an estimated average for the entire anatomical region being assessed. The degree of spongiosis and chronic degeneration (neuronal loss and gliosis) was also graded using a similar semiquantitative method: − none, + minimal, ++ mild, +++ moderate, ++++ severe. In addition to semiquantitative assessment, neuronal intranuclear inclusions (NII) were more accurately quantified by manually counting the total number of NII in 20 adjacent 20× microscopic fields in layer II of the most severely affected neocortical region. Senile plaque and neurofibrillary tangle pathology were scored according to CERAD and Braak staging methods, respectively (Braak and Braak, 1991; Mirra et al., 1991).
Statistical analysis
Principal component analysis was first applied to the entire group of FTLD-U cases to identify the most significant pathological features that explain the variation. Descriptive statistics were then used to further characterize the individual clinical subgroups. χ2-test was employed to compare the semiquantitative pathological features among the various groups, and Kendall's tau was used to assess trends. As multiple comparisons were made, a Bonferroni corrected significant P-value would be 0.000625. This would likely be over-conservative as several pathological variables were strongly correlated based on the initial principal component analysis. Given that five components explain ∼80% of the variance, and we were comparing two main groups (mutation-positive versus mutation-negative), we adopted P-values <0.005 as significant, and considered P-values between 0.05 and 0.005 to be suggestive only.
Results
Neuropathology of cases with proven PGRN mutations (Cases 1–13)
Gross findings
In the 12 autopsy cases with confirmed PGRN mutations, gross examination of the post-mortem brain specimen showed some degree of cerebral atrophy in all cases, with the weight ranging from 940 to 1250 g (mean = 1073 g). In 11/12 cases, there was severe atrophy of the frontal lobes with mild-to-moderate involvement of the temporal and parietal lobes. The exception was Case 12 in which the parietal lobes showed severe degeneration (left > right) while the frontal lobes were moderately atrophic. In 8/12, the head of the caudate nucleus was noticeably shrunken an in 2/12 cases there was loss of pigmentation from the substantia nigra.
Microscopic features (general)
Microscopic examination demonstrated non-specific chronic degenerative changes of neuronal loss, gliosis and microglial activation. The frontal neocortex was most severely affected in all cases with the exception of Case 12 (parietal > frontal). There was some degeneration of the caudate and putamen in all cases with 8/12 being at least moderately affected. Loss of pigmented neurons from the substantia nigra was variable (3 mild, 7 moderate, 1 severe, 2 not available). In the cases where the thalamus was available for examination, there was moderate or severe degeneration of the medial nuclear group in 6/8. In 8/12 cases there was selective loss of pyramidal neurons from the CA1 sector of the hippocampus and in 4 of these, the degeneration was severe enough to warrant the additional diagnosis of hippocampal sclerosis. Cerebrovascular disease, in the form of chronic infarction or subdural haematoma, was found in 5/12 cases. One patient (Case 6) also had multifocal demyelination, consistent with chronic multiple sclerosis.
Immunohistochemistry with antibodies against amyloid-β, tau, α-synuclein and neurofilament proteins, failed to demonstrate any pathology beyond that expected in patients of this age group (Mackenzie, 1994; Braak and Braak, 1997). Neuritic plaques were present in 2 cases (1 sparse, 1 moderate) (CERAD) and neurofibrillary tangles were present in the limbic structures of the mesial temporal lobe in 4/12 (2 Stage I/VI, 1 Stage II/VI, 1 Stage III/VI). Only one case (no. 13) had both neuritic senile plaques (moderate) and neurofibrillary tangles (Stage I/VI).
Microscopic features (specific)
Cases with mutations had a highly consistent pattern of pathology (Table 2). The most characteristic feature was the presence of ub-ir lentiform NII in the neocortex and striatum (Fig. 1). Based on their dimensions, oval and round NII were interpreted as representing lentiform inclusions cut in tangential or cross-section, respectively (Fig. 1C). Although the number of NII was never numerous, they were never difficult to find in sections of affected neocortex and striatum. In neocortex, their frequency ranged from 1/1 to 1/4 20× microscopic fields (mean = 1/2 20× fields). NII were less consistently found in other anatomic locations, such as hippocampus and brainstem (Fig. 1D). In addition to NII, the neocortex in all cases showed moderate-to-severe superficial laminar spongiosis, chronic degenerative changes, ub-ir neurites and ub-ir NCI (Fig. 2A–C). The cortical NCI were mostly well-defined and solid in appearance with the majority being either curved or oval in shape (Fig. 2C). NCI with an annular shape and those with a more granular consistency were infrequent. Although their number was more variable, NCI were always present in hippocampal dentate granule cells (Fig. 2D and E). In this location, they were almost exclusively granular; some were quite ill-defined and diffuse while others had the appearance of a dense aggregate of coarse granules (Fig. 2E). Smooth-contoured, solid-appearing NCI were only rarely seen in the hippocampus, in a few cases. Occasionally, one or two lentiform NII were identified in dentate granule cells (Fig. 1D). In the striatum, the degree of degeneration varied, however, in all cases (with the exception of Case 2), there were numerous ub-ir neurites (Fig. 2F). NCI in the striatum were less numerous than in the neocortex and could be either solid or granular. In only one case (Case 3) was pathology identified in LMN, with moderate loss of spinal anterior horn cells and a single filamentous ub-ir NCI identified in the hypoglossal nucleus.
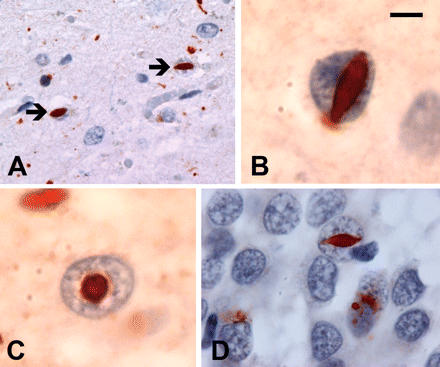
(A and B) All cases with proven PGRN mutations had lentiform ub-ir neuronal intranuclear inclusions (NII) in the neocortex and striatum (arrows). (C) Round NII likely represent lentiform inclusions viewed in cross-section. (D) NII were occasionally seen in other regions such as the hippocampal dentate granule cells. Ubiquitin immunohistochemistry. (A) Scale bar = 40 μm, (B and C) scale bar = 4 μm and (D) scale bar = 20 μm.
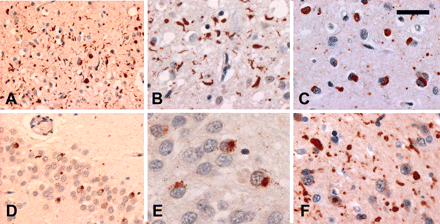
In addition to NII, all cases with PGRN mutations showed similar pathological changes (Table 2). (A–C) In layer II of the frontal neocortex, there were moderate-to-numerous ub-ir neurites (B) and neuronal cytoplasmic inclusions (NCI) (C). Most NCI were well-defined and had a curved or oval shape (C). (D and E) NCI in hippocampal dentate granule cells were variable in number and most had a granular appearance. (F) Numerous ub-ir neurites were present in the striatum. Ubiquitin immunohistochemistry. (A) Scale bar = 66 μm, (B and C) scale bar = 33 μm, (D) scale bar = 50 μm, (E) scale bar = 13 μm and (F) scale bar = 33 μm.
Neuropathological findings in cases with FTLD-U
| Neocortex | Hippocampus | Striatum | LMN | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spng | Deg | Neur | NCI solid | NCI gran | NCI total | NII | NCI solid | NCI gran | NCI total | NII | Deg | Neur | NCI solid | NCI gran | NCI total | NII | Deg | NCI | |
| PGRN mut + (n = 13) | |||||||||||||||||||
| 1 | +++ | +++ | +++ | +++ | − | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| 2 | ++++ | +++ | +++ | +++ | + | +++ | ++ | − | ++ | ++ | − | + | ++ | − | + | + | + | − | − |
| 3 | +++ | +++ | ++++ | +++ | + | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | ++ | Single |
| 4 | ++++ | ++++ | ++++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | + | + | + | ++ | − | − |
| 5 | +++ | +++ | ++++ | +++ | + | +++ | ++ | + | ++ | ++ | − | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 6 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | ++ | ++ | ++ | − | − |
| 7 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | na | na |
| 8 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | − | ++ | ++++ | ++ | ++ | +++ | ++ | na | na |
| 9 | ++++ | ++++ | ++++ | ++++ | + | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | + | ++ | ++ | − | − |
| 10† | na | na | ++++ | +++ | ++ | +++ | ++ | na | na | na | na | na | na | na | na | na | na | na | na |
| 11 | ++++ | ++++ | ++++ | +++ | + | ++++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | + | ++ | ++ | − | − |
| 12 | +++ | ++++ | ++++ | +++ | ++ | +++ | ++ | − | +++ | +++ | + | ++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 13 | ++++ | ++++ | +++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| Mean | ++++ | +++ | ++++ | +++ | + | +++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| PGRN mut − (n = 5) | |||||||||||||||||||
| Mean | +++ | + | ++ | ++ | ++ | +++ | − | ++ | ++++ | ++++ | − | + | ++ | ++ | + | ++ | − | + | ++ |
| Range | +/++++ | +/++ | +/+++ | +/++++ | +/++ | +/++++ | − | ++/+++ | ++/++++ | ++++ | − | −/++ | +/+++ | +/++ | +/++ | +/++ | − | −/+++ | −/+++ |
| P-value | * | ** | ** | * | * | ** | * | * | * | * | ** | ** | * | ||||||
| NII + PGRN na (n = 8) | |||||||||||||||||||
| Mean | ++ | ++ | +++ | ++++ | + | +++ | ++ | + | +++ | +++ | + | + | ++++ | ++ | ++ | +++ | + | ++ | + |
| Range | +/+++ | +/++++ | ++/++++ | ++/++++ | −/+++ | ++/++++ | +/++ | −/+++ | +/++++ | +/++++ | −/++ | −/+++ | +++/++++ | +/+++ | +/++++ | ++/++++ | −/++ | −/+++ | −/++ |
| P-value | * | * | * | * | * | * | * | * | |||||||||||
| FTD no NII PGRN na (n = 10) | |||||||||||||||||||
| Mean | +++ | +++ | +++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | ++ | ++ | ++ | ++ | ++ | − | − | + |
| Range | +/++++ | +/++++ | +/++++ | +/++ | −/++ | +/+++ | − | +/++++ | +/++++ | +/++++ | − | −/+++ | ++/+++ | −/++++ | +/+++ | +/++++ | − | −/+ | −/++ |
| P-value | * | ** | ** | * | * | * | * | ||||||||||||
| MND-d no NII PGRN na (n = 13) | |||||||||||||||||||
| Mean | +++ | ++ | ++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | + | ++ | + | + | ++ | − | ++ | +++ |
| Range | −/++++ | +/++++ | +/+++ | +/++++ | −/+++ | −/++++ | − | +/++++ | −/++++ | +/++++ | − | −/+++ | +/++++ | −/++++ | −/++ | −/++++ | − | +/+++ | −/+++ |
| P-value | ** | ** | * | * | ** | * | * | ** | * | ** | ** | ||||||||
| Neocortex | Hippocampus | Striatum | LMN | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spng | Deg | Neur | NCI solid | NCI gran | NCI total | NII | NCI solid | NCI gran | NCI total | NII | Deg | Neur | NCI solid | NCI gran | NCI total | NII | Deg | NCI | |
| PGRN mut + (n = 13) | |||||||||||||||||||
| 1 | +++ | +++ | +++ | +++ | − | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| 2 | ++++ | +++ | +++ | +++ | + | +++ | ++ | − | ++ | ++ | − | + | ++ | − | + | + | + | − | − |
| 3 | +++ | +++ | ++++ | +++ | + | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | ++ | Single |
| 4 | ++++ | ++++ | ++++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | + | + | + | ++ | − | − |
| 5 | +++ | +++ | ++++ | +++ | + | +++ | ++ | + | ++ | ++ | − | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 6 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | ++ | ++ | ++ | − | − |
| 7 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | na | na |
| 8 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | − | ++ | ++++ | ++ | ++ | +++ | ++ | na | na |
| 9 | ++++ | ++++ | ++++ | ++++ | + | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | + | ++ | ++ | − | − |
| 10† | na | na | ++++ | +++ | ++ | +++ | ++ | na | na | na | na | na | na | na | na | na | na | na | na |
| 11 | ++++ | ++++ | ++++ | +++ | + | ++++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | + | ++ | ++ | − | − |
| 12 | +++ | ++++ | ++++ | +++ | ++ | +++ | ++ | − | +++ | +++ | + | ++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 13 | ++++ | ++++ | +++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| Mean | ++++ | +++ | ++++ | +++ | + | +++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| PGRN mut − (n = 5) | |||||||||||||||||||
| Mean | +++ | + | ++ | ++ | ++ | +++ | − | ++ | ++++ | ++++ | − | + | ++ | ++ | + | ++ | − | + | ++ |
| Range | +/++++ | +/++ | +/+++ | +/++++ | +/++ | +/++++ | − | ++/+++ | ++/++++ | ++++ | − | −/++ | +/+++ | +/++ | +/++ | +/++ | − | −/+++ | −/+++ |
| P-value | * | ** | ** | * | * | ** | * | * | * | * | ** | ** | * | ||||||
| NII + PGRN na (n = 8) | |||||||||||||||||||
| Mean | ++ | ++ | +++ | ++++ | + | +++ | ++ | + | +++ | +++ | + | + | ++++ | ++ | ++ | +++ | + | ++ | + |
| Range | +/+++ | +/++++ | ++/++++ | ++/++++ | −/+++ | ++/++++ | +/++ | −/+++ | +/++++ | +/++++ | −/++ | −/+++ | +++/++++ | +/+++ | +/++++ | ++/++++ | −/++ | −/+++ | −/++ |
| P-value | * | * | * | * | * | * | * | * | |||||||||||
| FTD no NII PGRN na (n = 10) | |||||||||||||||||||
| Mean | +++ | +++ | +++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | ++ | ++ | ++ | ++ | ++ | − | − | + |
| Range | +/++++ | +/++++ | +/++++ | +/++ | −/++ | +/+++ | − | +/++++ | +/++++ | +/++++ | − | −/+++ | ++/+++ | −/++++ | +/+++ | +/++++ | − | −/+ | −/++ |
| P-value | * | ** | ** | * | * | * | * | ||||||||||||
| MND-d no NII PGRN na (n = 13) | |||||||||||||||||||
| Mean | +++ | ++ | ++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | + | ++ | + | + | ++ | − | ++ | +++ |
| Range | −/++++ | +/++++ | +/+++ | +/++++ | −/+++ | −/++++ | − | +/++++ | −/++++ | +/++++ | − | −/+++ | +/++++ | −/++++ | −/++ | −/++++ | − | +/+++ | −/+++ |
| P-value | ** | ** | * | * | ** | * | * | ** | * | ** | ** | ||||||||
PGRN, progranulin gene; mut, mutation status; na, not available; FTD, frontotemporal dementia; MND-d, motor neuron disease with dementia; spng, spongiosis; deg, degeneration; neur, ub-ir neurites; NCI, ub-ir neuronal cytoplasmic inclusions; gran, granular; NII, ub-ir neuronal intranuclear inclusions; LMN, lower motor neurons. Grading: −, none; +, rare/minimal; ++, mild; +++, moderate; ++++, numerous/severe. P-values: compared to group with proven PGRN mutations.
*P < 0.05; **P < 0.005; †Patient had neurosurgical biopsy only.
Neuropathological findings in cases with FTLD-U
| Neocortex | Hippocampus | Striatum | LMN | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spng | Deg | Neur | NCI solid | NCI gran | NCI total | NII | NCI solid | NCI gran | NCI total | NII | Deg | Neur | NCI solid | NCI gran | NCI total | NII | Deg | NCI | |
| PGRN mut + (n = 13) | |||||||||||||||||||
| 1 | +++ | +++ | +++ | +++ | − | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| 2 | ++++ | +++ | +++ | +++ | + | +++ | ++ | − | ++ | ++ | − | + | ++ | − | + | + | + | − | − |
| 3 | +++ | +++ | ++++ | +++ | + | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | ++ | Single |
| 4 | ++++ | ++++ | ++++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | + | + | + | ++ | − | − |
| 5 | +++ | +++ | ++++ | +++ | + | +++ | ++ | + | ++ | ++ | − | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 6 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | ++ | ++ | ++ | − | − |
| 7 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | na | na |
| 8 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | − | ++ | ++++ | ++ | ++ | +++ | ++ | na | na |
| 9 | ++++ | ++++ | ++++ | ++++ | + | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | + | ++ | ++ | − | − |
| 10† | na | na | ++++ | +++ | ++ | +++ | ++ | na | na | na | na | na | na | na | na | na | na | na | na |
| 11 | ++++ | ++++ | ++++ | +++ | + | ++++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | + | ++ | ++ | − | − |
| 12 | +++ | ++++ | ++++ | +++ | ++ | +++ | ++ | − | +++ | +++ | + | ++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 13 | ++++ | ++++ | +++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| Mean | ++++ | +++ | ++++ | +++ | + | +++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| PGRN mut − (n = 5) | |||||||||||||||||||
| Mean | +++ | + | ++ | ++ | ++ | +++ | − | ++ | ++++ | ++++ | − | + | ++ | ++ | + | ++ | − | + | ++ |
| Range | +/++++ | +/++ | +/+++ | +/++++ | +/++ | +/++++ | − | ++/+++ | ++/++++ | ++++ | − | −/++ | +/+++ | +/++ | +/++ | +/++ | − | −/+++ | −/+++ |
| P-value | * | ** | ** | * | * | ** | * | * | * | * | ** | ** | * | ||||||
| NII + PGRN na (n = 8) | |||||||||||||||||||
| Mean | ++ | ++ | +++ | ++++ | + | +++ | ++ | + | +++ | +++ | + | + | ++++ | ++ | ++ | +++ | + | ++ | + |
| Range | +/+++ | +/++++ | ++/++++ | ++/++++ | −/+++ | ++/++++ | +/++ | −/+++ | +/++++ | +/++++ | −/++ | −/+++ | +++/++++ | +/+++ | +/++++ | ++/++++ | −/++ | −/+++ | −/++ |
| P-value | * | * | * | * | * | * | * | * | |||||||||||
| FTD no NII PGRN na (n = 10) | |||||||||||||||||||
| Mean | +++ | +++ | +++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | ++ | ++ | ++ | ++ | ++ | − | − | + |
| Range | +/++++ | +/++++ | +/++++ | +/++ | −/++ | +/+++ | − | +/++++ | +/++++ | +/++++ | − | −/+++ | ++/+++ | −/++++ | +/+++ | +/++++ | − | −/+ | −/++ |
| P-value | * | ** | ** | * | * | * | * | ||||||||||||
| MND-d no NII PGRN na (n = 13) | |||||||||||||||||||
| Mean | +++ | ++ | ++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | + | ++ | + | + | ++ | − | ++ | +++ |
| Range | −/++++ | +/++++ | +/+++ | +/++++ | −/+++ | −/++++ | − | +/++++ | −/++++ | +/++++ | − | −/+++ | +/++++ | −/++++ | −/++ | −/++++ | − | +/+++ | −/+++ |
| P-value | ** | ** | * | * | ** | * | * | ** | * | ** | ** | ||||||||
| Neocortex | Hippocampus | Striatum | LMN | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spng | Deg | Neur | NCI solid | NCI gran | NCI total | NII | NCI solid | NCI gran | NCI total | NII | Deg | Neur | NCI solid | NCI gran | NCI total | NII | Deg | NCI | |
| PGRN mut + (n = 13) | |||||||||||||||||||
| 1 | +++ | +++ | +++ | +++ | − | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| 2 | ++++ | +++ | +++ | +++ | + | +++ | ++ | − | ++ | ++ | − | + | ++ | − | + | + | + | − | − |
| 3 | +++ | +++ | ++++ | +++ | + | +++ | ++ | − | + | + | − | +++ | ++++ | + | ++ | ++ | ++ | ++ | Single |
| 4 | ++++ | ++++ | ++++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | + | + | + | ++ | − | − |
| 5 | +++ | +++ | ++++ | +++ | + | +++ | ++ | + | ++ | ++ | − | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 6 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | ++ | ++ | ++ | − | − |
| 7 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | na | na |
| 8 | ++++ | +++ | ++++ | ++++ | − | ++++ | ++ | + | +++ | +++ | − | ++ | ++++ | ++ | ++ | +++ | ++ | na | na |
| 9 | ++++ | ++++ | ++++ | ++++ | + | ++++ | ++ | − | +++ | +++ | + | ++ | ++++ | + | + | ++ | ++ | − | − |
| 10† | na | na | ++++ | +++ | ++ | +++ | ++ | na | na | na | na | na | na | na | na | na | na | na | na |
| 11 | ++++ | ++++ | ++++ | +++ | + | ++++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | + | ++ | ++ | − | − |
| 12 | +++ | ++++ | ++++ | +++ | ++ | +++ | ++ | − | +++ | +++ | + | ++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| 13 | ++++ | ++++ | +++ | +++ | − | +++ | ++ | − | ++++ | ++++ | + | +++ | ++++ | ++ | ++ | +++ | ++ | − | − |
| Mean | ++++ | +++ | ++++ | +++ | + | +++ | ++ | − | +++ | +++ | + | +++ | ++++ | + | ++ | ++ | ++ | − | − |
| PGRN mut − (n = 5) | |||||||||||||||||||
| Mean | +++ | + | ++ | ++ | ++ | +++ | − | ++ | ++++ | ++++ | − | + | ++ | ++ | + | ++ | − | + | ++ |
| Range | +/++++ | +/++ | +/+++ | +/++++ | +/++ | +/++++ | − | ++/+++ | ++/++++ | ++++ | − | −/++ | +/+++ | +/++ | +/++ | +/++ | − | −/+++ | −/+++ |
| P-value | * | ** | ** | * | * | ** | * | * | * | * | ** | ** | * | ||||||
| NII + PGRN na (n = 8) | |||||||||||||||||||
| Mean | ++ | ++ | +++ | ++++ | + | +++ | ++ | + | +++ | +++ | + | + | ++++ | ++ | ++ | +++ | + | ++ | + |
| Range | +/+++ | +/++++ | ++/++++ | ++/++++ | −/+++ | ++/++++ | +/++ | −/+++ | +/++++ | +/++++ | −/++ | −/+++ | +++/++++ | +/+++ | +/++++ | ++/++++ | −/++ | −/+++ | −/++ |
| P-value | * | * | * | * | * | * | * | * | |||||||||||
| FTD no NII PGRN na (n = 10) | |||||||||||||||||||
| Mean | +++ | +++ | +++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | ++ | ++ | ++ | ++ | ++ | − | − | + |
| Range | +/++++ | +/++++ | +/++++ | +/++ | −/++ | +/+++ | − | +/++++ | +/++++ | +/++++ | − | −/+++ | ++/+++ | −/++++ | +/+++ | +/++++ | − | −/+ | −/++ |
| P-value | * | ** | ** | * | * | * | * | ||||||||||||
| MND-d no NII PGRN na (n = 13) | |||||||||||||||||||
| Mean | +++ | ++ | ++ | ++ | + | ++ | − | ++ | +++ | ++++ | − | + | ++ | + | + | ++ | − | ++ | +++ |
| Range | −/++++ | +/++++ | +/+++ | +/++++ | −/+++ | −/++++ | − | +/++++ | −/++++ | +/++++ | − | −/+++ | +/++++ | −/++++ | −/++ | −/++++ | − | +/+++ | −/+++ |
| P-value | ** | ** | * | * | ** | * | * | ** | * | ** | ** | ||||||||
PGRN, progranulin gene; mut, mutation status; na, not available; FTD, frontotemporal dementia; MND-d, motor neuron disease with dementia; spng, spongiosis; deg, degeneration; neur, ub-ir neurites; NCI, ub-ir neuronal cytoplasmic inclusions; gran, granular; NII, ub-ir neuronal intranuclear inclusions; LMN, lower motor neurons. Grading: −, none; +, rare/minimal; ++, mild; +++, moderate; ++++, numerous/severe. P-values: compared to group with proven PGRN mutations.
*P < 0.05; **P < 0.005; †Patient had neurosurgical biopsy only.
Immunohistochemistry for PGRN demonstrated diffuse cytoplasmic reactivity in a subset of neocortical and hippocampal pyramidal neurons and intense staining of activated microglial cells (Fig. 3). No PGRN-positive neurites, NCI or NII were identified. There was no immunoreactivity of the NII with the 1C2 antibody.
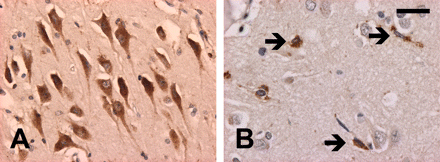
In all cases, PGRN immunohistochemistry demonstrated granular cytoplasmic staining in a subset of pyramidal neurons, including the hippocampus (A) and in activated microglial cells (arrows) (B). In cases with PGRN mutations, there was no staining of neurites or neuronal inclusions in the neocortex (B). PGRN immunohistochemistry. (A and B) Scale bar = 23 μm.
Mutation-positive cases versus mutation-negative cases
The five familial FTLD-U cases in which PGRN mutations had been excluded differ significantly from those with mutations. The neocortical pathology was generally milder with less degeneration (P = 0.001), fewer neurites (P = 0.003) and no cortical NII (P < 0.0005). Striatal neurites were significantly fewer (P = 0.004) and there were no striatal NII (P < 0.0005). Solid NCI were more frequent in the hippocampus (P = 0.005) (Fig. 4A). In those cases that had a clinical presentation of both MND and dementia, there was mild-to-moderate loss of anterior horn cells and NCI in LMN (Fig. 4B).
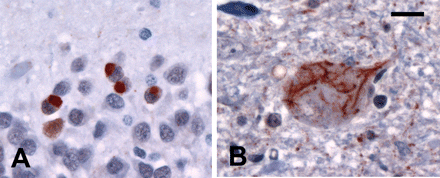
In cases without mutations or NII, hippocampal NCI more often had a solid, rather than granular, appearance (A). All cases with clinical MND-dementia had characteristic ub-ir cytoplasmic inclusions in lower motor neurons (B). Ubiquitin immunohistochemistry. (A and B) Scale bar = 15 μm.
Mutation-positive cases versus other cases with NII
Eight cases in which genetic analysis was not available (4 clinical FTD, 4 clinical MND-dementia) were also found to have NII with a similar morphology and distribution as those in cases with mutations. Although there was a trend for the neocortical pathology to be slightly less severe, the overall pattern of pathology was similar to those with proven mutations.
Mutation-positive cases versus cases of FTLD-U without NII
Once cases with NII had been excluded, none of the remaining FTLD-U cases showed the same pattern of pathology as those with proven PGRN mutations. Cases with clinical FTD (n = 10) had significantly fewer neocortical NCI (P < 0.0005), a trend to less severe striatal pathology and more frequent solid NCI in the hippocampus (P = 0.009). Cases with clinical MND-dementia (n = 13) had significantly less neocortical degeneration (P = 0.002), fewer neocortical and striatal neurites (P < 0.0005) and more frequent solid NCI in hippocampus (P = 0.001). In addition, all cases with clinical MND had a loss of LMN with ub-ir NCI in some of the remaining LMN (P < 0.0005).
Discussion
This study provides the first detailed description of the neuropathology in patients with FTD and identified PGRN mutations. All our patients with proven mutations showed a very similar pattern of neurodegeneration and ub-ir pathology. The lack of significant variation among cases, despite them having mutations in different regions of the PGRN gene, is consistent with our current hypothesis that all the mutations that cause FTD, do so by a common (haploinsufficiency) mechanism (Baker et al., 2006; Cruts et al., 2006). The absence of any PGRN-immunoreactive pathological inclusions, further supports our previous evidence that the mutant PGRN mRNA, in all the cases we examined, is destroyed (via a nonsense mediated decay mechanism) (Marquat, 2004; Baker et al., 2006) without there being any significant production of mutant protein (Baker et al., 2006).
Other than FTLD-U, the only other type of neuropathology that seemed to be more common in cases with PGRN mutations was the presence of hippocampal sclerosis, found in 33%. The reason for an apparent association between these two conditions is uncertain but has been reported previously (Hatanpaa et al., 2004; Josephs et al., 2004). Perhaps the fact that hippocampal pyramidal neurons normally express very high levels of PGRN puts them at increased risk of neurodegeneration in these cases (Fig. 3) (Daniel et al., 2000). Cerebrovascular disease was also common (42%) but it is hard to know if this is beyond what would be expected for age. We found no evidence of an increased age-related frequency or severity of either neuritic senile plaques or neurofibrillary tangles (Mackenzie, 1994; Braak and Braak, 1997), perhaps making it unlikely that PGRN mutations play a role in Alzheimer's disease pathogenesis.
Cases of familial FTD in which PGRN mutations had been excluded showed significant differences compared with mutation-positive cases. Even though some of those without mutations had clinical MND (in addition to FTD), the milder extramotor pathology could not be explained by the duration of their dementia (mean = 5 years, range = 2–8 years), since it was similar to those with mutations (Table 1). In addition, mild-to-moderate numbers of solid NCI were present in dentate granule cells in all the mutation-negative cases but were only rarely found in those with mutations. However, the most consistent difference was the presence of lentiform NII, which were present in all cases with mutations but not found in any of the mutation-negative cases. Subsequent to our initial publication of PGRN mutations in FTD (Baker et al., 2006), many additional cases have been identified and, to our knowledge, all of those in which pathology is available show NII (Gass et al., 2006). It is also worth noting that several other chromosome 17q21 linked FTLD-U families reported in the literature are also known to have NII (Kertesz et al., 2000; Rosso et al., 2001; Rademakers et al., 2002; Bennusi et al., 2004). These findings suggest that NII are a highly sensitive pathologic marker for PGRN mutations and their demonstration may be a way of identifying cases and families that should undergo genetic screening.
We also found similar lentiform NII in eight cases in which it was not possible to screen for mutations (due to lack of a source of DNA or inability to obtain patient consent for genetic analysis). In addition to NII, these cases had an overall pattern of neurodegeneration and ub-ir pathology that was similar to those with proven mutations, suggesting the possibility that these other cases with NII might also have PGRN mutations. Interestingly, only 4/8 cases with NII and unknown genetic status had an obvious family history of disease. One previous study also reported NII in rare cases of apparently sporadic FTD (Bigio et al., 2004). Although some of these cases might have unrecognized relevant family history or an inherited mutation with incomplete penetrance, it is also possible that some cases of sporadic FTD could be due to either spontaneous mutations in PGRN or some other abnormality of PGRN protein production or metabolism.
Cases with PGRN mutations showed significant differences when compared with our other remaining FTLD-U cases, without NII. Although we have previously suggested that the FTLD-U pathology in cases with clinical FTD and with MND-dementia is similar (Mackenzie and Feldman, 2005), other studies have suggested the two groups may be distinct (Katsuse and Dickson, 2005); therefore, we did separate comparisons of each clinical group with the PGRN mutation group. Not only was each of these groups significantly different from the mutation-positive group, but there was no single case of either FTD or MND-dementia without NII that showed the same pattern of pathology as the cases with mutations. Specifically, no other case showed the combination of numerous ub-ir neurites in the neocortex and striatum and a predominance of granular NCI in the hippocampal dentate granule cells. These findings suggest that, even though NII may be the most characteristic pathological feature of cases with PGRN mutations, the overall pattern of ub-ir pathology is also distinct.
Special mention should be made regarding the rarity of ub-ir NCI in LMN in our cases with PGRN mutations. Such inclusions are felt to be a highly sensitive marker for MND (Lowe, 1994) and were found in 15/17 MND cases in this study. However, several previous studies have noted similar LMN inclusions in a proportion of FTLD-U cases with a clinical history of FTD without motor features (Mackenzie and Feldman, 2005), suggesting that FTD, MND-dementia and classical MND may represent a clinical spectrum of disease with a common underlying pathology. In this study, once cases with PGRN mutations or NII were excluded, 38% of the remaining cases with FTD only, had LMN NCI. Unfortunately, the spinal cord was available for examination in only one of our mutation-positive cases (Case 3). Nonetheless, thorough examination of this case and of brainstem motor nuclei in the remaining 12 cases, identified only a single filamentous ub-ir NCI, in the hypoglossal nucleus, in one case. Not only does the scarcity of LMN pathology in the mutation-positive cases further distinguish them from the rest of the FTLD-U spectrum, but it may be an indication that PGRN mutations are unlikely to be a major cause of familial MND.
Ub-ir lentiform NII were first described in detail by Woulfe et al. (2001) in a small number of cases of clinical FTD with FTLD-U pathology (Woulfe et al., 2001). Similar inclusions were mentioned briefly in the pathological description of two FTD families linked to chromosome 17q21–22 (Rosso et al., 2001; Rademakers et al., 2002;). A previous review of our material found NII only in cases of familial FTLD-U with an autosomal dominant pattern of inheritance and we proposed they might be a pathological marker for a specific genetic cause of FTD (Mackenzie and Feldman, 2003; Mackenzie et al., 2006a). Although the current study confirms that lentiform NII are a highly sensitive marker for PGRN mutations, we now know that they are not entirely specific. The rare autosomal dominant syndrome of ‘inclusion body myopathy associated with Paget disease of bone and FTD’ (IMPFD) is caused by mutations in the gene that encodes the valosin-containing protein (Watts et al., 2004). The neuropathology of IMPFD has recently been described as a unique subtype of FTLD-U that includes numerous lentiform NII (Forman et al., 2006). Although the NII have a similar morphology and anatomical distribution, they are much more numerous in IMPFD than in other cases of familial FTLD-U. Other differences include more widespread cortical pathology in IMPFD (including the occipital lobe), less involvement of subcortical structures and significantly fewer NCI, particularly in the hippocampus.
The fact that lentiform NII have not been reported in other conditions, but are a consistent feature of two distinct FTD syndromes caused by different genetic abnormalities, is intriguing and suggests that PGRN and VCP may lead to neurodegeneration through some common mechanism. The nature of this process and the significance of NII remain unknown, however, we have previously reported that the ub-ir NII in one of our families (UBC-17) were also immunoreactive for both promyelocytic leukaemia protein and small ubiquitin modifier-1 (Mackenzie et al., 2006a). These findings suggest the NII might contribute to neuronal degeneration by causing proteosomal dysfunction and by disrupting the normal function of the nuclear body.
Although the identification of PGRN mutations has been an important step in unravelling the pathogenesis of FTD, it leaves many crucial questions unanswered and raises several new issues. All of the cases with PGRN mutations that have been identified thus far have clinical FTD and FTLD-U pathology. However, this strong clinical/pathological/genetic correlation may simply reflect biases in how we have initially collected cases to study. It still needs to be determined whether or not PGRN mutations are responsible for other clinical or pathological conditions (such as other types of dementia or MND) and whether polymorphisms in PGRN are risk factors for sporadic FTD or other conditions. We still do not know the identity of the ubiquitinated protein that accumulates in these cases and whether or not it is the same in all cases of FTLD-U. Finally, clarifying the specific role of PGRN in neurodegeneration will be crucial to developing rational therapeutic approaches.
In conclusion, null mutations in PGRN have now been established as an important cause of familial FTD. The presence of this second FTD gene, just 1.7 Mb from MAPT explains why many families with FTD show linkage to chromosome 17q21 but do not have an identifiable MAPT mutation. The neuropathology associated with FTD caused by PRGN mutations is a unique subtype of FTLD-U in which the most characteristic feature is the presence of ub-ir lentiform NII.
Immunohistochemistry was performed by Julie Chow (Department of Pathology, University of British Columbia) and the technical staff in the immunohistochemistry laboratory at Vancouver General Hospital. Tissue samples were generously provided by Dr Chris Dunham, Dr Arthur Clark and Dr Bernadette Curry (Foothills Hospital, Calgary, Canada), Dr John Woulfe (Ottawa Civic Hospital, Ottawa, Canada), Dr John Rossiter (Queen's University, Kingston, Canada) and Dr Catherine Joachim (The Radcliffe Infirmary, Oxford, UK). I.R.A.M., G.R.H., C.L., E.D. and H.H.F. supported by the Canadian Institutes of Health Research (grant 74580). M.B., J.G., A.C. and M.H. were supported by the National Institute of Health (grants P01 AG017216, R01 AG026251), the Mayo Clinic ADRC (P50 AG16574) and the Mayo Clinic Research Foundation. S.P.B. was supported by the UK Medical Research Council Motor Neuron Disease Association. R.R. supported by the Fund for Scientific Research, Flanders.


{kind=link}
{kind=link}
{kind=link}
{kind=link}