




Abstract
Frontotemporal lobar degeneration (FTLD) refers to a focal, non-Alzheimer form of cerebral degeneration that encompasses the distinct clinical syndromes of frontotemporal dementia (FTD), progressive non-fluent aphasia (PNFA) and semantic dementia. Some patients show tau-based pathological changes and in familial cases mutations have been identified in the microtubule-associated protein tau gene (MAPT) on chromosome 17q21. However, many cases are tau-negative, showing instead ubiquitin-immunoreactive (UBQ-ir) neuronal cytoplasmic inclusions and neurites, and in some familial cases UBQ-ir neuronal intranuclear inclusions of a lentiform appearance. Very recently, mutations have been identified in familial cases in the progranulin (PGRN) gene, also on chromosome 17q21. Clinical, pathological and molecular diversity within FTLD highlights the importance of careful examination of clinical-pathological-genetic relationships. This paper reports, for the first time, a clinico-pathological investigation of two FTLD families with PGRN mutations, and compares the clinical characteristics with those of patients studied in the department with MAPT mutations. The clinical profile associated with PGRN mutations constituted, in some patients, a prototypical picture of FTD and in others one of PNFA, both profiles occurring within the same family. Patients with PGRN mutations exhibited phonological deficits, whereas in patients with MAPT mutations language abnormalities, when present in addition to the prominent behavioural disorder, take the form of semantic disturbance. The findings provide compelling evidence for the link between FTD and PNFA, while raising the possibility of identifiable clinical differences between FTLD patients with MAPT and PGRN mutations.
Abbreviations
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- MAPT
microtubule-associated protein tau gene
- PGRN
progranulin
- PNFA
progressive non-fluent aphasia
- SD
semantic dementia
- UBQ-ir
ubiquitin-immunoreactive
Introduction
Frontotemporal lobar degeneration (FTLD) is a descriptive term that refers to a clinically and pathologically heterogeneous group of non-Alzheimer forms of dementia with disease onset often before 65 years of age (Snowden et al., 1996a; Neary et al., 1998). The different clinical subtypes of FTLD include (i) frontotemporal dementia (FTD), in which behavioural and personality changes predominate, (ii) semantic dementia (SD), characterized by loss of conceptual knowledge and (iii) progressive non-fluent aphasia (PNFA), in which the presenting characteristic is impaired language expression. Each of these clinical forms of FTLD likely reflects differing topographical distributions of similar underlying pathologies.
FTLD is highly familial with up to 40% cases showing a pattern of inheritance consistent with autosomal dominant transmission of disease (Rosso et al., 2003; Neary et al., 2005). About 20% of such familial cases have mutations in the microtubule-associated protein tau (MAPT) gene, located on chromosome 17q21 (reviewed in Mann, 2005). In these cases, aggregated hyperphosphorylated tau proteins are deposited in the brain in the form of intraneuronal neurofibrillary tangles or Pick bodies (Taniguchi et al., 2004; Shi et al., 2005). Nonetheless, as many as 60% FTLD cases do not show such pathological changes in tau in their brains, but display ubiquitin-immunoreactive (UBQ-ir) neuronal cytoplasmic inclusions and neuritic changes in the cerebral cortex and the hippocampus (Shi et al., 2005; Mackenzie et al., 2006b). In certain FTLD families with autosomal dominant transmission of disease (Rosso et al., 2001; Rademakers et al., 2002; Froelich Fabre et al., 2003; Mackenzie et al., 2006a), UBQ-ir neuronal intranuclear inclusions of a lentiform or ‘cat’s eye’ appearance (Woulfe et al., 2001; Mackenzie and Feldman, 2003) are seen within neurons in the same regions of the cerebral cortex and hippocampus where the neuronal cytoplasmic inclusions and neuritic changes are present. Although these families had been previously reported (Foster et al., 1997) to show linkage to the same region on chromosome 17q21 that contains MAPT, it had not subsequently been possible to identify any pathogenic MAPT mutation(s) (Basun et al., 1997; Froelich et al., 1997; Rosso et al., 2001; Rademakers et al., 2002; Froelich Fabre et al., 2003; Mackenzie et al., 2006a).
However, it has very recently been shown that most of these families are associated with mutations within the progranulin (PGRN) gene (Baker et al., 2006; Cruts et al., 2006), this being located just 1.7 Mb upstream of MAPT. Several genetic alterations have been identified, including various insertion/deletion mutations resulting in frameshifts, mutations in initiation codons and nonsense mutations (Baker et al., 2006; Cruts et al., 2006). In one Manchester family, known as F337 the Q486X mutation was present, whereas in the other family (F53), the Q130SfsX124 mutation was present (Baker et al., 2006). Baker et al. (2006) report two other frameshift mutations (C31LfsX34 and T382SfsX29), three other nonsense mutations (Q125X, W386X and R418X), and a mutation in the 5′-splice site of exon 8 (IVS8+1G→A). In Cruts et al., (2006), the most common mutation present was within a large Belgian family (known as DR8), where there was a G–C transversion in intron 0 at a position +5 relative to first non-coding exon 0 (IVS0+5G→C). Four other mutations, one nonsense (Q125X), two involving frameshifts (P127RfsX1 and A237WfsX3) and one G→A transition destroying Met 1 translation initiation codon in exon were detected in this latter study. In every instance, the PGRN mutations are predicted to cause premature termination of the coding sequence, creating null alleles with the mutant RNAs likely being degraded by nonsense mediated decay. These data imply that the neurodegenerative process in these FTLD patients stems from loss of functional PGRN (i.e. haploinsufficiency).
The clinical characteristics of patients showing mutations in PGRN have not hitherto been described. In particular, it is not known whether PGRN mutations are associated with a specific clinical phenotype of FTLD and whether this differs from that of patients showing mutations in MAPT. In this report, we present the clinical, neuropsychological and pathological characteristics of two families with FTLD (F53 and F337) associated with a mutation in PGRN (Baker et al., 2006) ascertained within the Greater Manchester region of UK. Approval for clinical, pathological and molecular components of the study was granted by the local Research Ethics Committee.
Material and methods
Clinical methods
In family F337 the proband and one of her affected sisters had been examined in the department, and in family F53 the proband alone. For each of these patients the clinical history was elicited using a structured proforma that addresses systematically domains of cognition and behaviour, ensuring consistency of data ascertainment. Cognitive evaluation was carried out using the Manchester Neuropsychological Assessment (Neary et al., 1986; Thompson et al., 2005), which provides a profile of performance in the domains of language, calculation, perception, spatial functions, praxis, memory and executive abilities. The instrument has been found valuable in predicting regional areas of dysfunction on functional imaging (Neary et al., 1987; Talbot et al., 1995, 1998) and in differentiating forms of dementia (Thompson et al., 2005). The instrument was supplemented by standard, published neuropsychological tests. Structural and functional imaging was undertaken in both probands. Clinical information about affected family members, not seen in the department, was obtained through access to their hospital files and from relatives' reports.
Pathological methods
In F337, the entire brain of the proband, apart from some small samples of right frontal and temporal cortex, and cerebellum which were frozen at −80°C for genetic analysis, was fixed by immersion in 10% buffered formalin for 4 weeks prior to examination and sectioning. For F53, the left cerebral hemisphere, cerebellum and brainstem of the proband were also fixed in 10% buffered formalin, whereas the right cerebral hemisphere was again frozen at −80°C. Representative tissue blocks were cut from the cerebral cortex and other regions to include, frontal, temporal, cingulate, parietal and occipital cortex, hippocampus, amygdala, basal ganglia, substantia nigra, brainstem and cerebellar cortex. Tissue blocks were routinely processed into paraffin wax and sections cut at thickness of 6 μm. Sections were stained by routine neurohistological methods, including Weigert's haematoxylin-eosin and Luxol fast blue, and immunohistochemically using a standard avidin–biotin–peroxidase method (Taniguchi et al., 2004; Shi et al., 2005). For the immunodetection of tau, AT8 (Ser 202/Thr 205) (1 : 200), AT180 (Thr 231) (1 : 200), AT270 (Thr 181) (1 : 200) (all from Innogenetics, Belgium), CP13 (Ser 202) and PHF-1 (Ser396/404) (gift of Dr P. Davies, 1 : 2000 and 1 : 1000, respectively), 12E8 (Ser 262) (gift of Dr P. Seubert, 1 : 200) and the 3R (RD3, de Silva et al., 2006, 1 : 3000) and 4R (ET3, gift of Dr P Davies, 1 : 100) tau specific monoclonal antibodies were used. Sections stained by the latter antibodies required pressure-cooker pretreatment (5 min in sodium citrate buffer, pH 6.0) before incubation in primary antibody as described elsewhere (de Silva et al., 2006; Shiarli et al., 2006). Further immunoreactions used three PGRN primary antibodies (R&D Systems, Minneapolis, MN, 1 : 500 and N-terminus, acro granin, N-19, 1 : 100 and C-terminus, acrogranin, S-15, 1 : 100, both from Santa Cruz Biotechnology, Santa Cruz, CA) which recognize all regions of PGRN protein including the amino and carboxyl termini (Santa Cruz antibodies) and the full-length recombinant human PGRN protein (R&D antibody). Sections for PGRN immunostaining were also microwaved for antigen retrieval for 5 min in sodium citrate buffer, pH 6.0, before incubation in primary antibody as described elsewhere (Shiarli et al., 2006). Other immunostaining included 4G8 anti amyloid β protein (Signet Labs, Denham, MA, 1:2000 with 5 min formic acid pretreatment), ubiquitin (Dako, Ely, UK 1 : 750), GFAP (Sigma, 1 : 750) and α-synuclein (Chemicon, 1 : 1000 with 4 min trypsin pretreatment) antibodies. All primary incubations were performed overnight at 4°C, except those for ubiquitin which were performed using Ventana automated staining system, as described elsewhere (Mackenzie et al., 2006b).
Results
Family history
Affected individuals were identified in three generations (Fig. 1A). These comprised the proband, two of her elder sisters, her mother and maternal grandmother. The proband and one sister developed prominent problems in both language and behaviour, whereas the other sister presented with aphasia without evident behavioural change. Her mother and maternal grandmother both showed prominent behavioural symptoms without aphasia.
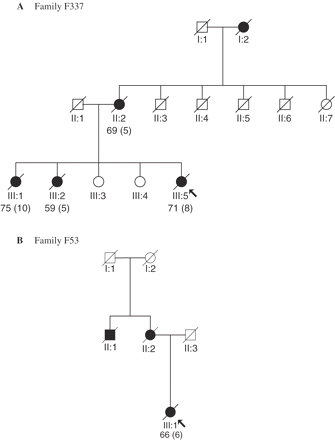
Family trees for (A) family F337 and (B) family F53. Filled symbols indicate that the individual was definitely affected; arrows indicate the pedigree position of the proband. Age at death (with the duration of illness where known) is indicated.
Clinical findings of proband (Patient III.5, Fig. 1A)
This woman was referred at the age of 65 years with a 2-year history of changes in her language and behaviour. She had become less expressive in speech and she had difficulty finding the appropriate word. However, she did not make word errors and it was believed that her comprehension was retained. She continued to read, but wrote less. Her husband had taken over the control of the family finances, so he could not comment on her arithmetical skills.
Her personality had changed. She lacked motivation and required considerable prompting in carrying out household and social chores, and she could no longer prepare meals as before. Her self-care had declined and she had to be prompted in matters of hygiene and grooming. She expressed no concern or insight into her altered mental state. She did not exhibit ritualistic behaviours or change in her eating habits.
She was thought to be a little forgetful. However, she had no visual symptoms and was able to negotiate the environment and find her way without difficulty. There had never been any confusional episodes, and she was not hallucinated or deluded.
Aside from longstanding deafness inherited from her father, she had no physical symptoms, and there was no relevant previous medical history.
Neurological examination
On clinical examination she had a fatuous affect and her clothes were stained. Voluntary eye movements were slightly reduced on vertical gaze and there were bilateral grasp reflexes. The remainder of the neurological examination was normal.
Cognitive evaluation
The neuropsychological evaluation revealed prominent deficits in the domains of language and executive functions.
Her spontaneous speech was mildly hesitant and stuttering and she made occasional phonemic paraphasias. However, sentences were grammatically correct. She performed well on a series of tasks that required her to complete sentences from a phrase, or generate sentences from a one-word cue (Snowden et al., 1996b). No abnormalities were detected in language comprehension at a word or sentence level, and her interpretation of metaphor and proverb was appropriately abstract. She achieved normal scores on the ‘pyramids and palm trees test’ of semantic association (Howard and Patterson, 1992), which examines word understanding and the test of reception of grammar (Bishop, 1989) that examines sentence comprehension.
However, she could not follow sequential commands. She had a reduced forward digit span of four digits and she made sequencing errors. She made both sequencing and phonological errors in repeating words and phrases. Naming performance was within normal limits. She scored 16/30 on the difficult graded naming test (McKenna and Warrington, 1983). However, she made phonological errors, which she spontaneously corrected. There were also occasional verbal substitutions, but no frank semantic category errors (e.g. horse for kangaroo). Her category fluency performance was within normal limits (17 animals in 1 min), but letter fluency was reduced (6 F-, 3 A- and 10 S-words). She made phonological errors in reading. She had difficulty spelling, both oral and written. By contrast, she could calculate, solving accurately both mental and written additions and subtractions.
No abnormalities were detected in perception, spatial functions or praxis. She identified line drawings and famous faces. She localized objects and traced a maze. She passed spatial subtests of the visual-object and spatial perception battery (Warrington and James, 1991). She demonstrated gestures and action pantomimes to verbal command and copied non-representational hand postures. Her drawings were spatially accurate, but there was error of detail, suggesting failure of self-monitoring. Her memory performance was patchy, being poorest for verbal tasks. She was broadly oriented in time and place and she could give an account of personally relevant events. However, she performed poorly on a story recall test. Her performance on executive tasks was mildly impaired. She succeeded in abstracting three alternative sorting rules in the Weigl's block sorting test (De Renzi et al., 1966). However, she made order errors in a picture sequencing task. In a design fluency task (Jones-Gotman and Milner, 1977) she showed poor generation of ideas and made rule-violation errors.
The cognitive profile was of impaired language together with mild executive failure.
Neuroimaging
A CT scan of the brain carried out two years after onset of symptoms, revealed cerebral atrophy particularly marked in the left cerebral hemisphere. A subsequent MR scan confirmed the asymmetrical distribution of atrophy (Fig. 2). A SPECT scan (Fig. 2) demonstrated poor perfusion in both frontotemporal and anterior parietal lobes, more pronounced on the left side.
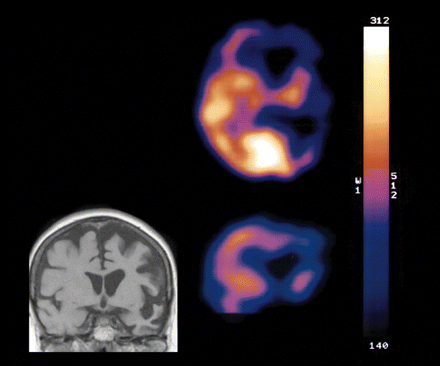
MR (coronal section) and SPECT scan (transaxial and coronal sections) of F337 proband, showing asymmetrical atrophy and impaired tracer uptake in left hemisphere.
Progression
Over the following 4 years she became increasingly apathetic and lacking in motivation. She became emotionally blunted and was doubly incontinent without concern. She initially developed eating fads, which her relatives described as ‘picky’, but later, she ate excessively and indiscriminately and became overweight. She developed stereotypic movements of the left leg, which would swing to and fro, and from side to side, repetitively. She began to walk more slowly, with a stooped posture. There was increasing loss of the use of her right arm and hand. Three years after her initial referral she was sufficiently incapacitated to require 24 h care in the activities of daily living and the use of a wheelchair.
Neurological examination at that time revealed an expressionless face with further reduction of voluntary upward gaze. She was profoundly rigid, especially in the right arm and leg, but without any change in muscular power. There were bilateral grasp reflexes. The right arm and hand were held in flexion, and there was profound loss of dexterity of the right hand. Her state of akinesia and rigidity was not responsive to the administration of l-dopa.
There was a progressive decline in her language. Her speech output became increasingly economical and unelaborated and she could no longer generate or complete unfinished sentences. Her responses to questions became limited to monosyllables. Initially, she could recite on request well-rehearsed verbal series, such as the months of the year, and name common objects, albeit with phonemic and perseverative errors. However, 4 years after her initial referral she was totally mute. No speech sounds could be elicited either through repetition, reading or series speech tasks, although she was noted to mouth words. During the time that she was formally testable her word comprehension, measured by a four-choice word–picture matching test, remained well preserved (40/40 correct). However, ultimately her performance was compromised by profound inattention and perseverative responses. She was unable to perform a sentence ordering task, involving arrangement of five words to form a sentence.
In addition to the salient language and executive deficits, she increasingly neglected to use her right arm. Her copies of gestures (e.g. salute, foot tapping) were substantially more degraded with her right hand and foot than her left. After 6 years, from the onset of symptoms, no communication was possible either through speech or gesture. She died at the age of 71, 8 years after onset of symptoms.
Clinical diagnosis
The clinical diagnosis was of FTLD. She exhibited a mixed clinical syndrome, which combined the apathetic behavioural syndrome of FTD, the language disorder of PNFA and neurological features of parkinsonism.
Neuropathological findings of proband
At post-mortem, the whole brain weighed 863 g. External examination showed a grossly asymmetrical atrophy, this being severe within the left cerebral hemisphere, though the right cerebral hemisphere was much less atrophic. The major cerebral arteries appeared normal. Atrophy within the left cerebral hemisphere involved all cortical regions from frontal pole to occipital pole. The cerebellum and brainstem were small but without external abnormality.
On coronal serial section, the lateral ventricle of the left cerebral hemisphere was moderately enlarged throughout, though the right lateral ventricle was less dilated. The temporal horn extension was likewise enlarged on the left side, but not on the right side. In the left cerebral hemisphere, there was gross atrophy of the frontal (Fig. 3A) and anterior parietal cortex. The inferior and middle temporal gyri were grossly atrophied though the superior temporal gyrus was relatively preserved. The left posterior parietal cortex and occipital cortex were only moderately atrophic. The deep white matter of the left cerebral hemisphere showed severe loss of myelin, having a brownish colouration, being soft and ‘rubbery’. The distinction between grey and white matter was poorly maintained. The right cerebral hemisphere showed much less atrophy, with the white matter being well myelinated and the distinction between grey and white matter well maintained. There was considerable atrophy of the caudate nucleus and putamen on the left side, but with better preservation on the right side of the brain. The globus pallidus and thalamus were also moderately atrophic on the left side of the brain, though both were normal on the right. The corpus callosum was thinned at all levels. There was considerable atrophy of the parahippocampal gyrus on the left side, though the hippocampus and amygdala were better preserved. The right hippocampus, parahippocampal gyrus and amygdala appeared normal. The left substantia nigra showed complete loss of neuromelanin pigment, whereas the right substantia nigra was moderately well pigmented. The rest of the midbrain, brainstem and cerebellum, on both sides, appeared normal. There were no cerebrovascular changes of significance.
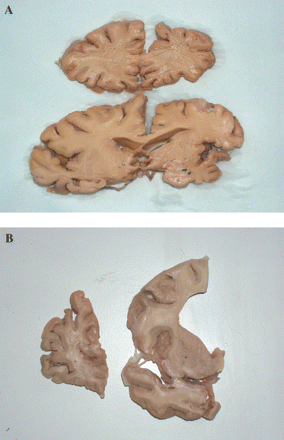
(A) Shows coronal sections of brain of proband of F337 at level of frontal lobe (upper) and mid temporal lobe (lower). Note the markedly asymmetric atrophy affecting left side of brain (shown on right) preferentially. The white matter is softened and fragmentary on left side, both within frontal and temporal lobes. The hippocampus and amygdala are mildly atrophied, as are basal ganglia. (B) Shows coronal sections of brain of proband of F53 showing severe atrophy of frontal lobe and moderate atrophy of temporal lobe and basal ganglia. Again, the deep white matter of the frontal lobe is soft and fragmentary.
The left frontal, temporal and anterior parietal cortex all showed a virtually complete loss of nerve cells from layers 2, 3 and 5, with extensive microvacuolation and collapse of the neuropil leading to complete loss of cytoarchitecture. There was extensive loss of axons and myelin from the deep white matter with some preservation of the U-fibres. Swollen cells (ballooned neurons) were not present. There was widespread reactive astrocytosis throughout all cortical layers. Similar, but less severe, changes were seen in the superior temporal gyrus, and the posterior parietal and occipitoparietal cortices, where superficial laminar microvacuolation was present. The same kind of histopathological changes, but much less severe, were seen throughout the right cerebral cortex, even in frontal and temporal lobes these were relatively mild. There was virtually complete loss of nerve cells from areas CA1 and subiculum of the hippocampus on both left and right sides, with extensive reactive astrocytosis. Other regions of the hippocampus showed only little nerve cell loss, though there was a moderate reactive astrocytosis throughout all regions. The caudate nucleus and putamen showed extensive astrocytosis. There was considerable loss of nerve cells from the substantia nigra with a moderate reactive astrocytosis, but no Lewy bodies were present. The cerebellum and dentate nucleus were histologically normal. Many small arteries within the deep white matter of the cerebral cortex showed a moderate arteriosclerosis, but without tissue infarction.
Immunohistochemical findings
There were a moderate number of UBQ-ir intracytoplasmic neuronal inclusions and neurites within layer 2 of the left and right frontal and anterior parietal cortex, though both of these changes were numerous within the temporal cortex (Fig. 4A). A moderate number (∼5 per section) of ‘cat's eye’ or lentiform neuronal intranuclear inclusions were seen in small neurons of layer 2 of frontal and temporal cortex (Fig. 4B). In the hippocampus, there was a moderate number of intracytoplasmic inclusions within granule cells of the dentate gyrus (Fig. 4C), but no intranuclear inclusions were seen. Immunostaining with PGRN antibodies showed occasional immunopositive microglial cells, and some immunopositive dendrites of cerebral cortical pyramidal cells, though the UBQ-ir intracytoplasmic neuronal inclusions, neurites and neuronal intranuclear inclusions were all unstained (data not shown).
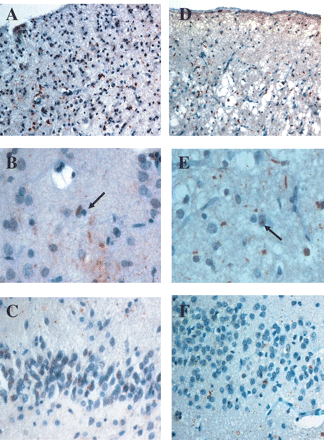
Ubiquitin pathology in the probands of F337 (A–C) and F53 (D–F). In both patients there is only a moderate number of ubiquitin-positive neurites and neuronal cytoplamic inclusions in temporal cortex (A and D). Occasional neuronal nuclear inclusions (arrowed) are present in both (B and E). There are only few, rather granular, ubiquitin cytoplasmic inclusions in granule cells of the dentate gyrus of the hippocampus (C and F). Ubiquitin immunoreactivity–haematoxylin, magnification ×200 (A and D), ×600 (B and E), ×300 (C and F).
No deposits of tau protein, in the form of neurofibrillary tangles, Pick bodies or amorphous tau deposits within nerve cells, or glial cells, were seen in any part of the brain using any of the tau antibodies employed. Immunostaining with 4G8 antibody revealed a mild deposition of amyloid β protein within anterior parietal, posterior parietal, posterior temporal and occipital cortices, mostly in the form of diffuse amyloid plaques though occasional cored plaques were present, particularly in posterior parietal and occipital regions. The extent of amyloid deposition was greater in right cerebral cortical regions of the brain. Amyloid deposits were absent or rare in both left and right frontal and anterior temporal cortex. Occasional diffuse plaques were present within the grey matter surrounding the dentate gyrus of the hippocampus on both the left and right sides of the brain. No amyloid deposits were present within basal ganglia regions or within the cerebellum. No α-synuclein-immunoreactive changes were seen.
Pathological diagnosis
The pathological diagnosis was of FTLD, associated with markedly asymmetric atrophy of the left hemisphere and ubiquitin-type histology.
Clinical findings of proband's eldest sister (Patient III:1, Fig. 1A)
This woman was referred at the age of 67 years with a 2-year history of difficulty in language and forgetfulness. She was acutely aware of these difficulties and showed appropriate frustration and concern, believing that her symptoms could neither be sufficiently explained by anxiety caused due to the stresses within the family nor by her awareness that both her mother and sister had died of a dementing illness. She was aware that at retirement from her business at the age of 65 years, the finances were in disarray.
She described progressive difficulties in naming, but with preservation of her powers of comprehension, reading and writing. She felt that her powers of calculation were slightly impaired. She described some forgetfulness, with a tendency to mislay objects, and in particular she had difficulty remembering people's names. She had no visual symptoms and was fully proficient in all the activities of daily living, including driving. She had taken up voluntary unpaid work in a café, seemingly without difficulty. She continued to take full responsibility for her family, despite irritability and anxiety, engendered by difficulties with her daughters and husband. She had no physical symptoms. In the past she had been treated for mild hypertension.
Neurological examination
The clinical neurological examination revealed reduction of voluntary upward gaze. There was slight increase in tone in the right arm, but no weakness. The remainder of the neurological examination was normal.
Neuropsychological evaluation
The prominent problem was in language expression. Her speech output was empty, lacking in substantives, reflecting word retrieval difficulty. She made word and occasional sound-based errors, but no frank semantic category errors. There was incorrect use of prepositions. She had a reduced repetition span of four digits forwards. Repetition of word sequences and phrases elicited phonological errors. The naming difficulty noted in conversational speech was mirrored by profound impairment on formal naming tests. On the relatively undemanding Boston Naming Test (Kaplan et al., 1983) she named only 9/60 items. Errors were typically circumlocutions, descriptions or gestural demonstrations (e.g. volcano—‘Etna—it goes right up’, dart—‘throw it in’, wreath—‘in December’). There were no semantic errors. By contrast to her anomia, comprehension was well preserved at a single word level. However, she had difficulty understanding complex syntax. She could read prose passages, write a sentence and spell common words aloud accurately. She had no difficulty carrying out mental and written calculations. She showed no impairment in visual perception, spatial functions or praxis. She identified line drawings of objects and famous faces, although could not retrieve their names. She conveyed gesture and action pantomimes accurately. She copied non-representational hand postures and motor sequences. She reproduced line drawings of objects and abstract figures, block designs and match-stick designs, including mirror reversals. Her memory was poor for verbal material, but preserved for visual material. She could reproduce drawings of abstract figures after a delay. Her performance on a forced-choice picture recognition test was normal. She performed normally on executive tasks that do not make verbal demands: the modified version of the Wisconsin Card Sorting Test (Nelson, 1976) and the Design Fluency Test (Jones-Gotman and Milner, 1977). However, verbal fluency was somewhat reduced: 11 animals and 7 F-words in 1 min.
The picture was of a relatively circumscribed aphasic disorder, characterized chiefly by anomia.
Progression
Her anomia increased over the next 2 years and her speech output became reduced to monosyllables and stereotyped utterances. She was thereafter lost to follow-up. She died in a nursing home at the age of 75, 10 years after onset of symptoms. No post-mortem examination was undertaken.
Clinical diagnosis
The clinical diagnosis was of PNFA, associated with mild parkinsonism.
Characteristics of proband's maternal grandmother (Patient I:2, Fig. 1A)
This woman died in her early 70s after being an invalid for several years, with severe neuritis and muscular weakness, and slow deterioration in her mental condition. She was described as talkative and would provide a running commentary on activities outside her window. She tended to fabricate events. At no time was she reported to have become mute.
Characteristics of proband's mother (Patient II:2, Fig. 1A)
This woman began to show behavioural changes at ∼64 years. She, like her mother, fantasized about people and animals outside her window. Her behaviour was socially inappropriate, and she would appear in company with her underwear around her ankles. She tended to wander. She died at the age of 69 years, 5 years after onset of symptoms. At no time was she reported to have become mute.
Characteristics of proband's second sister (Patient III:2, Fig. 1A)
This woman became increasingly withdrawn and apathetic at the age of 54. She neglected her appearance and became ill-kempt and obese. She wandered and would become lost. Her speech output became progressively reduced, and ultimately she became mute. She became incontinent and unable to use her left arm, which showed increased tone and pathological reflexes. She died aged 59 years, 5 years after symptom onset. An autopsy was carried out and said to suggest Pick's disease.
Family history
Three affected family members were identified (Fig. 1B): the proband, her mother and maternal uncle. The proband's mother had become demented in middle age and it was commented that she had exhibited behavioural changes similar to those of her daughter. Her uncle had moved away to Canada so that the precise nature of his dementing illness was uncertain.
Clinical findings in proband (II.2, Fig. 1B)
Clinical history
This 62-year-old woman was referred with a 2-year history of change in behaviour, following her retirement from work. Her husband described the change in conduct, into which she had no insight. She had no physical or mental complaints, and was unaware of the purpose of the referral and consultation. Her husband described longstanding deafness of the right ear and a left sided cataract, but aside from slowing down a little, she had no obvious physical infirmity.
There had, however, been a radical change in her personality and behaviour. She had lost her motivation and had given up all interest or participation in household and social activities. She neglected her household chores and appeared unable to cook a meal. She expressed no concern about her appearance and could not be persuaded to wash regularly. Occasionally she became irritable and verbally aggressive. Her eating habits had changed and she ate excessively, gluttonously and continually searched for food. Accordingly, her weight had increased by two stones in 3 months.
Her prevailing lack of activity was interrupted by episodes of purposeless wandering, both in the home and out of doors. She would insist on retiring to bed at precisely 6 p.m. and then she would wake in the middle of the night and wander around the home. On shopping expeditions with her husband she would repeatedly wander off, but always found her way home without difficulty.
She was inattentive and forgetful and would often misplace her belongings in the home. No difficulties were noted in her language and she spoke fluently and without error. She could produce her signature and reckon change accurately. She had no visual problems and was able to identify and locate objects in the immediate environment and negotiate her way out of doors without difficulty. She had never become frankly confused and she was not thought to be hallucinated or deluded.
She lived with her husband who was under considerable stress because of her behaviour and insomnia.
Neurological examination
On clinical examination she was unkempt and her clothes were stained with food. She was obese. She had a bland facial expression, occasionally breaking into a fatuous grin and giggling. Aside from her longstanding right sided deafness and left cataract, the neurological examination was normal.
Cognitive evaluation
Evaluation of her cognitive abilities revealed a profound, yet circumscribed dysexecutive syndrome. Her speech output was economical and lacking in prosody. However, articulation and phonation were normal. There were no grammatical or paraphasic errors. No abnormalities were identified in comprehension at a single word or sentence level. However, her interpretation of metaphors and proverbs was concrete. Routine screening of repetition, naming, spelling, reading and writing abilities revealed no abnormalities. She had a normal digit span of eight digits forwards. Similarly, she showed no primary abnormalities in the realm of perception, spatial function or praxis. She recognized line drawings and identified famous faces. She traced a road map and located towns accurately on a map of Great Britain. She manipulated objects normally, copied non-representational hand postures accurately and conveyed gesture and action pantomimes to verbal command. Her copies of drawings showed preserved overall spatial configuration and relationship between elements. She could reproduce a cube accurately. Nevertheless, drawings were executed rapidly without checking, so that in copying complex designs there were omissions and errors of detail. Her memory performance had a ‘frontal’ quality. Her free recall of short stories immediately after reading them aloud was minimal. However, correct information could be elicited by directed questions and provision of multiple choice alternatives. She was oriented in time and place and could give an account of personally relevant events, suggesting an absence of severe clinical amnesia.
In contrast to her preserved performance on tests of language, perception, spatial skills and praxis her performance on executive tests was grossly abnormal. On the undemanding Weigl's blocks test (De Renzi et al., 1966) in which she was required to group coloured block shapes according to a common feature (shape, colour or motif), she identified uncued no sorting rules. When blocks were sorted for her she identified one rule only. Thereafter her responses were perseverative. On a picture sequencing task she altered pictures minimally from their original random order. Although she could describe the contents of individual pictures she made no attempt to integrate these elements into a logical, coherent narrative. In a verbal fluency task she produced only six animal words and no F-words in 1 min. She made concrete, perseverative and rule-violation errors. In the animal task she named her own cat and dog, Milly and Spot, and required coaxing to produce names of animals in the abstract. She then perseverated the same responses. In the F-word test she produced proper names even though explicitly requested not to do so.
The cognitive profile, together with her bland, fatuous affect and absence of concern for her performance, strongly indicated impaired frontal lobe function
Neuroimaging
A CT scan of the brain revealed cerebral atrophy, most pronounced in the frontal lobes. A SPECT scan indicated hypoperfusion of the frontal lobes bilaterally.
Progression
Eight months after her initial assessment, there had been a dramatic deterioration. Physically, she had slowed down, and become doubly incontinent, without embarrassment. She had become profoundly apathetic and inert, but still exhibited purposeless wandering. She remained gluttonous, actively searching for food, and had begun to cram food into her mouth, to the point of choking. She had become excessively sleepy during the day, but continued to wake and wander at night time.
Her spontaneous speech had become progressively attenuated and was confined to rare and brief utterances. However, she would repeat what was said to her perseveratively. No paraphasias had been noted, either semantic or phonological. She appeared to have no visual problems, and could still negotiate the home. She required 24 h care for the activities of daily living.
The neurological examination revealed rigidity of the limbs, without weakness, and all the movements were carried out slowly. She was beyond formal psychological evaluation. It was not possible to engage her attention and she wandered restlessly round the room. She was virtually mute although showed echolalia. She was also echopraxic.
Shortly after this assessment her husband found it impossible to care for her and she was admitted to residential care. She continued to deteriorate and died at the age of 66 years, 6 years after the onset of her symptoms.
Clinical diagnosis
The clinical diagnosis was of the apathetic form of FTD.
Neuropathological findings
At post-mortem, the whole brain weighed 1100 g. The major cerebral arteries appeared normal. The brain showed a moderate and slightly asymmetrical atrophy favouring the left side, affecting chiefly the frontal, anterior parietal and temporal cortex (inferior and middle temporal gyri), with relative sparing of the superior temporal gyrus, posterior parietal and occipital cortex. The cerebellum and brainstem showed no external abnormality.
On coronal serial section, the lateral ventricle was grossly enlarged anteriorly (Fig. 3B), but only moderately so at its posterior extent, though the temporal horn extension was only mildly enlarged. There was severe atrophy of the frontal, cingulate and anterior parietal cortex (Fig. 3B). The inferior and middle temporal gyri were also severely atrophied though the superior temporal gyrus was relatively preserved (Fig. 3B). The posterior parietal cortex and occipital cortex were only mildly atrophic. The deep white matter of the frontal (especially) and temporal cortex showed loss of myelin and was rather soft and fragmentary. There was moderate atrophy of the caudate nucleus and putamen, but the globus pallidus and thalamus were mildly atrophic. The corpus callosum was much thinned at all levels. There was moderate atrophy of the hippocampus, parahippocampal gyrus and amygdala. The substantia nigra was well pigmented. The rest of the midbrain, brainstem and cerebellum appeared normal. There were no cerebrovascular changes of significance.
The frontal, temporal and anterior parietal cortex showed severe loss of nerve cells from layers 2 and 3, with extensive microvacuolation. Neurons in layer 5 were shrunken. There was extensive loss of axons and myelin from the deep white matter with some preservation of the U-fibres. Swollen cells (ballooned neurons) were not present. There was widespread reactive astrocytosis, through most severely in subpial regions and at the junction of the grey and white matter. The orbitofrontal cortex was affected more severely than the convex cortex. Similar, but less severe, changes were seen in the superior temporal gyrus, and the posterior parietal and occipitoparietal cortices. There was virtually complete loss of nerve cells from areas CA1 and subiculum of the hippocampus, with extensive reactive astrocytosis. The caudate nucleus and putamen showed mild astrocytosis. There was mild loss of nerve cells from the substantia nigra, with slight astrocytosis, but no Lewy bodies were present. The cerebellum and dentate nucleus were histologically normal. Large arteries within the globus pallidus showed a moderate calcification.
Immunohistochemical findings
There were many UBQ-ir intracytoplasmic neuronal inclusions and neurites within layer 2 of the frontal and anterior parietal cortex (Fig. 4D), though these were only sparse within the temporal cortex. Occasional (i.e. 2–5 per section) ‘cat's eye’ or lentiform neuronal intranuclear inclusions were seen in small neurons of layer 2 (Fig. 4E) of frontal and temporal cortex. In the hippocampus, there was a moderate number of rather ‘granular’ intracytoplasmic inclusions within granule cells of the dentate gyrus (Fig. 4F), but no intranuclear inclusions were seen. Immunostaining with PGRN antibodies again showed occasional immunopositive microglial cells, and some immunopositive dendrites, but UBQ-ir intracytoplasmic neuronal inclusions, neurites and neuronal intranuclear inclusions were likewise unstained (data not shown).
No deposits of tau protein, in the form of neurofibrillary tangles, Pick bodies or amorphous tau deposits within nerve cells, or glial cells, were seen in any part of the brain using any of the tau antibodies employed, nor did immunostaining with 4G8 antibody reveal any deposits of amyloid β protein. No α-synuclein-immunoreactive changes were seen.
Pathological diagnosis
The pathological diagnosis was of FTLD with ubiquitin histology.
Molecular genetics
Molecular genetics revealed presence of Q130SfsX124 mutation (in F53) and Q486X mutation (in F337) in PGRN as described previously (Baker et al., 2006). No missense, or exon 10 splice site, mutation in MAPT was present in either proband. The apolipoprotein E (APOE) genotype was ɛ3/ɛ3 in both probands. There were no further DNA samples available from any other members of either family for analysis.
Discussion
In this report we have described the clinical, neuropsychological and neuropathological features of patients from two families with FTLD (F53 and F337) associated with mutations in PGRN (Baker et al., 2006). Although the probands from both families share similar underlying histopathological changes with UBQ-ir neuronal cytoplasmic and nuclear inclusions and neurites being present, the clinical characteristics and topographic distribution of the pathological changes differed. In the proband of family F337 these were concentrated in the left cerebral hemisphere producing a clinical and neuropsychological profile predominantly of progressive aphasia, whereas in that of family F53 the changes were bilateral, chiefly within the frontal lobes and a prototypical clinical picture of apathetic FTD was present. As the two families show different mutations in PGRN the question arises whether it is the actual mutation per se that is responsible for producing the variation in clinical phenotype or whether different genetic modifiers or other factors are involved which impose overriding effects in terms of clinical phenotype. Because all PGRN mutations are argued to have identical functional effects (Baker et al., 2006), the latter scenario seems more likely.
It is instructive in this regard that within the F337 family the clinical phenotype was not identical. In the proband, the dominant feature of language disturbance was combined with alterations in behaviour and loss of insight. By contrast, the proband's eldest sister presented with a more circumscribed aphasic disorder without notable behavioural change. She was insightful, frustrated and concerned. She continued to be independent in self-care and activities of daily living, despite her communication disorder. The proband's mother and grandmother, on the other hand, were reported to display behavioural change without language symptoms. These phenotypic variations provide support for the view that FTD and primary progressive aphasia are linked and part of the spectrum of clinical manifestations of FTLD (Neary et al., 1998), the different clinical phenotypes reflecting variations in topographical distribution of pathology (Neary et al., 1993).
How do the present patients compare with those with mutations in the MAPT gene? Direct comparisons are particularly pertinent with those patients with exon 10 +16 splice site mutations in the MAPT gene who have been studied by us (Hutton et al., 1998; Pickering-Brown et al., 2002), using identical assessment procedures. FTD associated with MAPT mutations has acquired the designation FTD with parkinsonism linked to chromosome 17 or FTDP-17 (Foster et al., 1997), on the grounds that parkinsonism is an intrinsic part of the clinical syndrome. Both probands with PGRN mutations described here developed parkinsonism, although this was a late rather than early feature. However, in our MAPT exon 10 +16 cases too parkinsonism developed late in the course and was not a presenting characteristic. Thus, from a neurological perspective there were no notable differences between MAPT and PGRN cases.
From a behavioural perspective the MAPT exon 10 +16 cases reported previously (Pickering-Brown et al., 2002) all presented with social disinhibition and purposeless overactivity, progressing gradually to apathy over the disease course, interpreted as reflecting a spread of pathology from orbital into medial and dorsolateral frontal cortex (Snowden et al., 1996a). However, other FTD patients exhibit an apathetic behavioural profile from the outset (Snowden et al., 1996a, 2001). In the F337 and F53 probands reported here the frontal lobe behavioural profile was of apathy from the onset of symptoms, raising the possibility of a behavioural profile distinct from that of MAPT exon 10 +16 tau cases. However, in family F337 second-hand reports of the mother and grandmother suggest that the presenting behavioural disorder may not have been of apathy, suggesting that strong conclusions cannot be drawn.
In patients with MAPT exon 10 +16 tau mutation the behavioural disorder was the dominant presenting symptom and remained the salient problem throughout the disease course. Yet, language abnormalities were elicited on formal testing in most cases. In all, this took the form of impaired naming and word comprehension in keeping with semantic impairment, and suggesting temporal lobe pathology. No patient showed evidence of phonological impairment.
The language disorder in family F337 is different in two respects. First, for both the proband (III.5) and her elder sister (III.1) the language disorder represented the dominant presenting symptom, and in the case of the sister it constituted a localized deficit, resulting in a clinical diagnosis of primary progressive aphasia. Second, the language disorder was qualitatively distinct. Both patients in family F337 made phonological, but not semantic errors in conversation and naming tasks. The anomic sister typically provided circumlocutions and functional descriptions for words that she could not retrieve. She was insightful and showed frustration into her naming difficulty. Both performed well on word comprehension tests. These features suggest that patients' language disorder arises at a phonological and lexical level and does not reflect an underlying loss of word semantics. With progression, there was an increasing loss of propositional language, similar to that found previously in association with frontal lobe degeneration (Snowden et al., 1996b). The inability to generate language and to perform sentence completion tests is in keeping with a ‘dynamic aphasia’ (Luria and Tsvetkova, 1968; Costello and Warrington, 1989), linked to frontal lobe pathology. The overall profile would suggest pathological involvement both of the frontal lobes and traditional perisylvian language areas of the left hemisphere.
In keeping with the clinical findings, the F337 proband showed a markedly asymmetric distribution of pathological change, extending throughout the language areas of the left hemisphere and affecting parietal as well as temporal neocortex. By contrast patients with the MAPT exon 10 +16 tau mutations (Pickering-Brown et al., 2002) have shown bilateral and relatively symmetrical atrophy of the frontal and temporal lobes, sometimes described as ‘knife-edge’.
The question arises whether observed differences in clinical profile hold true for other patients with different MAPT mutations. Among the MAPT mutations, there is good evidence that the clinical phenotype is variable (reviewed in Spillantini et al., 2000; Janssen et al., 2002; Rademakers et al., 2004; Richardson and Neary, 2006). Patients differ in the degree of parkinsonism. Occasionally patients show neurological signs suggestive of progressive supranuclear palsy or corticobasal degeneration. Nevertheless, the prominent presenting feature is almost invariably of the behavioural disorder of FTD and, as in our MAPT exon 10 +16 cases, this is typically of the overactive, disinhibited type. In keeping with the clinical syndrome, atrophy is typically reported to be frontotemporal and symmetrical in distribution, contrasting with the asymmetry seen in our PGRN mutation cases. In MAPT mutation cases alterations in language are commonly present. Their precise nature is not consistently specified. Nevertheless, descriptions, when available (e.g. Bird et al., 1999; Janssen et al., 2002; Boeve et al., 2005; Neumann et al., 2005) typically indicate a fluent speech, with word finding difficulties, and not the characteristic profile of PNFA. Thus, the profile of MAPT mutation cases, including those with other mutations than the exon 10 +16 mutation, is broadly consistent with that seen in our own MAPT exon 10 +16 mutation cases, and is different from that of the PGRN mutation cases reported here.
The identification of mutations associated with FTD and PNFA raises the question of the basis for the third principle syndrome associated with FTLD, namely SD (Snowden et al., 1989; Hodges et al., 1992). As noted above, semantic deficits may be elicited on neuropsychological examination in patients with MAPT exon 10 +16 mutations. Yet, this is invariably in the context of a severe behavioural disorder, and is associated with gross frontal as well as temporal lobe atrophy. In pure SD, by contrast, the semantic impairment predominates and is associated with severe yet relatively circumscribed temporal lobe atrophy. Although SD has also been associated with a ubiquitin rather than tau-based histology (Shi et al., 2005; Mackenzie et al., 2006b, 2006c), this is predominantly in the form of UBQ-ir neurites with few or no intraneuronal cytoplasmic inclusions and no neuronal intranuclear inclusions (Mackenzie et al., 2006c), clearly contrasting with the type of ubiquitin pathology found in the present patients.
PGRN is a 593 amino acid (68.5 kDa) multifunctional growth factor that is composed of seven and a half tandem repeats of a 12-cysteine granulin motif. As reported by Baker et al. (2006), immunostaining with PGRN antibodies failed to detect the ubiquitinated intracytoplasmic neuronal inclusions, neurites and neuronal intranuclear inclusions indicating that the disease mechanism does not cause accumulation of PGRN within these latter pathological lesions and the identity of the ubiquitinated protein is therefore still unclear. The role of PGRN within the human nervous system in general, and within FTLD in particular, remains unknown. In peripheral tissues, PGRN has been ascribed a role in wound repair, development and inflammation by activating signalling cascades that control cell cycle progression and cell motility (He and Bateman, 2003). PGRN also appears to stimulate the induction of other growth factors including vascular endothelial growth factor (Tangkeangsirisin and Serrero, 2004). Although partial loss of PGRN apparently results in an adult-onset neurodegenerative disease (viz FTLD), increased expression of PGRN has been linked to tumorigenesis (He and Bateman, 2003). These contrasting observations demonstrate the critical importance of PGRN, and the regulatory mechanisms that control its expression. The finding of PGRN mutations opens up the possibility of much greater understanding of the complex relationship between clinical and pathological phenotype and molecular substrate in FTLD. Since, the PGRN mutations impose a null allele and are postulated to lead to an insufficiency of PGRN protein, there is clear therapeutic potential to correct this, and alleviate symptomatology, through a simple replacement strategy.
Dr Pickering-Brown is the recipient of a Medical Research Council (MRC) New Investigator Award, and receives other funding from MRC and the Motor Neurone Disease Association.


{kind=link}
{kind=link}
{kind=link}
{kind=link}