




Abstract
Leakage of the blood–brain barrier (BBB) is associated with various neurological disorders, including temporal lobe epilepsy (TLE). However, it is not known whether alterations of the BBB occur during epileptogenesis and whether this can affect progression of epilepsy. We used both human and rat epileptic brain tissue and determined BBB permeability using various tracers and albumin immunocytochemistry. In addition, we studied the possible consequences of BBB opening in the rat for the subsequent progression of TLE. Albumin extravasation in human was prominent after status epilepticus (SE) in astrocytes and neurons, and also in hippocampus of TLE patients. Similarly, albumin and tracers were found in microglia, astrocytes and neurons of the rat. The BBB was permeable in rat limbic brain regions shortly after SE, but also in the latent and chronic epileptic phase. BBB permeability was positively correlated to seizure frequency in chronic epileptic rats. Artificial opening of the BBB by mannitol in the chronic epileptic phase induced a persistent increase in the number of seizures in the majority of rats. These findings indicate that BBB leakage occurs during epileptogenesis and the chronic epileptic phase and suggest that this can contribute to the progression of epilepsy.
Abbreviations
- BBB
blood–brain barrier
- FJB
fluoro-jade B
- TLE
temporal lobe epilepsy
- SE
status epilepticus
Introduction
Due to its unique structure, the blood–brain barrier (BBB) is capable of limiting the penetration of a variety of substances from the blood into the brain. The BBB plays an important role in the homeostasis and is generally seen as a defence mechanism that protects the brain against various molecules that may enter the BBB. The BBB is composed of endothelial cells which form a diffusion barrier, due to the presence of tight junctions that firmly connect endothelial cells (Kettenmann and Ransom, 2005). In addition to this, efflux transporters of the ATP binding cassette family (e.g. P-glycoprotein and multi-drug resistance-associated proteins), located at the luminal side of endothelial cells, may restrict further entry of substances into the brain. To provide the brain with essential nutrients and remove excreted substances, endothelial cells also contain numerous membrane transporters (for review seeLee et al., 2001) involved in the influx/efflux of essential substrates such as glucose, amino acids, electrolytes and nucleosides or removal of xenobiotics.
Due to the development of small molecular weight tracers that enter the damaged BBB, disruption was found to be associated with various neurological disorders such as migraine (Dreier et al., 2005), postconcussion syndrome (Korn et al., 2005), multiple sclerosis (Minagar and Alexander, 2003) and epilepsy (Roch et al., 2002; Ballabh et al., 2004; Neuwelt, 2004; Seiffert et al., 2004). BBB disruption has been shown both in human (Mihaly and Bozoky, 1984) as well as in animal studies after acute seizures (Nitsch and Klatzo, 1983; Zucker et al., 1983; Lassmann et al., 1984; Ruth, 1984; Saija et al., 1992; Pont et al., 1995; Ilbay et al., 2003; Leroy et al., 2003; Oztas et al., 2003) and has been associated with abnormal EEG patterns (Tomkins et al., 2001; Korn et al., 2005; Pavlovsky et al., 2005). Moreover, Friedman and colleagues showed that focal opening of the BBB by direct cortical application of albumin-containing solution can lead to the generation of an epileptic focus in rats (Seiffert et al., 2004). However, it is not known how the BBB integrity changes during epileptogenesis and whether alterations in BBB permeability can contribute to spontaneous seizure progression. To get more insight into the role of BBB disruption in epileptogenesis and progression of epilepsy, we determined BBB permeability in epileptic rats and humans and studied the possible consequences of a compromised BBB for the subsequent seizure progression. Since previous studies have shown that albumin can serve as an indicator of compromised BBB function in a variety of pathophysiological conditions (Cornford and Hyman, 1999; Seiffert et al., 2004), we used albumin and albumin-binding dyes to visualize BBB leakage.
Material and methods
Albumin immunoreactivity (IR) in human brain
To determine BBB permeability in human epileptic brain, albumin extravasation was studied by immunocytochemistry. Brain material was obtained from the files of the departments of neuropathology of the Academic Medical Center (University of Amsterdam). Patients underwent resection of the hippocampus (n = 6) for medically intractable epilepsy. To reduce metabolic injury, the fissural blood supply was kept intact until removal of the hippocampus. The surgical material is directly fixed after dissection and therefore in optimal condition. In addition, autopsy material was used of two epilepsy patients that died during an acute status epilepticus (SE). These patients had a long history of epilepsy (Table 1) and died before pharmacological treatment was started to stop the SE. Pathological examination excluded encephalitis or meningitis. This material was compared to normal-appearing hippocampi of five autopsy specimens from patients without history of seizures or other neurological diseases. Tissue was obtained and used in a manner compliant with the Declaration of Helsinki. Table 1 summarizes the clinical features of all patients. Brain tissue was fixed in 10% buffered formalin, paraffin embedded, sectioned at 6 μm and mounted on organosilane-coated slides (Sigma, St Louis, MO, USA). Two hippocampal sections of each patient were processed for immunocytochemistry. Sections were deparaffinated in xylene, rinsed in ethanol (100, 95 and 70%) and incubated for 20 min in 0.3% hydrogen peroxide diluted in methanol. Slides were then washed with phosphate-buffered saline (PBS; 10 mM, pH 7.4) and incubated overnight in anti-albumin (rabbit anti-human albumin; 1 : 20 000; DakoCytomation, Glostrup, Denmark) at 4°C. Hereafter, sections were washed in PBS and stained with a polymer based peroxidase immunocytochemistry detection kit (PowerVision Peroxidase system, ImmunoVision, Brisbane, CA, USA). After washing, sections were stained with 3,3′-diaminobenzidine tetrahydrochloride (50 mg DAB, Sigma-Aldrich, Zwijndrecht, The Netherlands) and 5 μl 30% hydrogen peroxide in a 10 ml solution of Tris–HCl. Sections were counterstained with haematoxylin, dehydrated in alcohol and xylene and coverslipped. Sections incubated without anti-albumin or with preimmune serum were essentially blank.
Summary of the clinical and neuropathological data of the patients with epilepsy
| Patient/age (year)/gender | Clinical/pathological diagnosis | Seizure frequency/months | Age at onset (year) | Duration epilepsy (year) | Seizure type | AEDs | Follow-up (year) | Engel class |
|---|---|---|---|---|---|---|---|---|
| 1/17/M | TLE/HS (W3) | <5 | 11 | 6 | CPS | PHT/CBZ/PB | 6 | I A |
| 2/21/M | TLE/HS (W3) | >20 | 2 | 19 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 3/19/F | TLE/HS (W3) | 10 | 6 | 13 | CPS | CBZ/PB/VPA | 1 | I A |
| 4/33/F | TLE/HS (W3) | 10–30 | 15 | 18 | CPS/SGS | CBZ/PB/VPA | 2 | I A |
| 5/27/M | TLE/HS (W3) | 10–20 | 10 | 17 | CPS | CBZ/PB/VPA | 3 | I A |
| 6/48/M | TLE/HS (W3) | 5–10 | 17 | 31 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 7/18/M* | TLE/HMEG | 30 | <1 | 18 | CPS/SGS/SE | CBZ/PB | — | — |
| 8/23/M* | Epilepsy/ND | 10–20 | 9 | 14 | CPS/SGS/SE | CBZ/LEV | — | — |
| Patient/age (year)/gender | Clinical/pathological diagnosis | Seizure frequency/months | Age at onset (year) | Duration epilepsy (year) | Seizure type | AEDs | Follow-up (year) | Engel class |
|---|---|---|---|---|---|---|---|---|
| 1/17/M | TLE/HS (W3) | <5 | 11 | 6 | CPS | PHT/CBZ/PB | 6 | I A |
| 2/21/M | TLE/HS (W3) | >20 | 2 | 19 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 3/19/F | TLE/HS (W3) | 10 | 6 | 13 | CPS | CBZ/PB/VPA | 1 | I A |
| 4/33/F | TLE/HS (W3) | 10–30 | 15 | 18 | CPS/SGS | CBZ/PB/VPA | 2 | I A |
| 5/27/M | TLE/HS (W3) | 10–20 | 10 | 17 | CPS | CBZ/PB/VPA | 3 | I A |
| 6/48/M | TLE/HS (W3) | 5–10 | 17 | 31 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 7/18/M* | TLE/HMEG | 30 | <1 | 18 | CPS/SGS/SE | CBZ/PB | — | — |
| 8/23/M* | Epilepsy/ND | 10–20 | 9 | 14 | CPS/SGS/SE | CBZ/LEV | — | — |
AEDs = antiepileptic drugs; CBZ = carbamazepine; CPS = complex partial seizures; HMEG = hemimegalencephaly; HS = hippocampal sclerosis; LEV = levetiracetam; ND = pathology not defined at autopsy; PB = phenobarbital; PHT = phenytoin; SE = status epilepticus; SGS = secondary generalized seizures; TLE = temporal lobe epilepsy; VPA = valproate; W = Wyler grading system.
*Autopsy patients.
Summary of the clinical and neuropathological data of the patients with epilepsy
| Patient/age (year)/gender | Clinical/pathological diagnosis | Seizure frequency/months | Age at onset (year) | Duration epilepsy (year) | Seizure type | AEDs | Follow-up (year) | Engel class |
|---|---|---|---|---|---|---|---|---|
| 1/17/M | TLE/HS (W3) | <5 | 11 | 6 | CPS | PHT/CBZ/PB | 6 | I A |
| 2/21/M | TLE/HS (W3) | >20 | 2 | 19 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 3/19/F | TLE/HS (W3) | 10 | 6 | 13 | CPS | CBZ/PB/VPA | 1 | I A |
| 4/33/F | TLE/HS (W3) | 10–30 | 15 | 18 | CPS/SGS | CBZ/PB/VPA | 2 | I A |
| 5/27/M | TLE/HS (W3) | 10–20 | 10 | 17 | CPS | CBZ/PB/VPA | 3 | I A |
| 6/48/M | TLE/HS (W3) | 5–10 | 17 | 31 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 7/18/M* | TLE/HMEG | 30 | <1 | 18 | CPS/SGS/SE | CBZ/PB | — | — |
| 8/23/M* | Epilepsy/ND | 10–20 | 9 | 14 | CPS/SGS/SE | CBZ/LEV | — | — |
| Patient/age (year)/gender | Clinical/pathological diagnosis | Seizure frequency/months | Age at onset (year) | Duration epilepsy (year) | Seizure type | AEDs | Follow-up (year) | Engel class |
|---|---|---|---|---|---|---|---|---|
| 1/17/M | TLE/HS (W3) | <5 | 11 | 6 | CPS | PHT/CBZ/PB | 6 | I A |
| 2/21/M | TLE/HS (W3) | >20 | 2 | 19 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 3/19/F | TLE/HS (W3) | 10 | 6 | 13 | CPS | CBZ/PB/VPA | 1 | I A |
| 4/33/F | TLE/HS (W3) | 10–30 | 15 | 18 | CPS/SGS | CBZ/PB/VPA | 2 | I A |
| 5/27/M | TLE/HS (W3) | 10–20 | 10 | 17 | CPS | CBZ/PB/VPA | 3 | I A |
| 6/48/M | TLE/HS (W3) | 5–10 | 17 | 31 | CPS/SGS | PHT/CBZ/PB | 1 | I A |
| 7/18/M* | TLE/HMEG | 30 | <1 | 18 | CPS/SGS/SE | CBZ/PB | — | — |
| 8/23/M* | Epilepsy/ND | 10–20 | 9 | 14 | CPS/SGS/SE | CBZ/LEV | — | — |
AEDs = antiepileptic drugs; CBZ = carbamazepine; CPS = complex partial seizures; HMEG = hemimegalencephaly; HS = hippocampal sclerosis; LEV = levetiracetam; ND = pathology not defined at autopsy; PB = phenobarbital; PHT = phenytoin; SE = status epilepticus; SGS = secondary generalized seizures; TLE = temporal lobe epilepsy; VPA = valproate; W = Wyler grading system.
*Autopsy patients.
Experimental animals
To evaluate whether changes of BBB permeability occurred during epileptogenesis, the SE rat model for temporal lobe epilepsy (TLE) was used (Gorter et al., 2001).
Adult male Sprague–Dawley rats (Harlan CPB Laboratories, Zeist, The Netherlands) weighing 400–550 g were housed individually in a controlled environment (21 ± 1°C; humidity 60%; lights on from 08:00 a.m. to 8:00 p.m.; food and water available ad libitum). The study was approved by the University Animal Welfare committee.
Electrode implantation
Rats were anaesthetized with ketamine (57 mg/kg; Alfasan, Woerden, The Netherlands) and xylazine (9 mg/kg; Bayer AG, Leverkusen, Germany) and placed in a stereotactic frame. In order to record hippocampal EEG, a pair of insulated stainless steel electrodes (70 μm wire diameter, tips were 0.8 mm apart) were implanted into the left dentate gyrus under electrophysiological control as previously described (Gorter et al., 2001). A pair of stimulation electrodes was implanted in the angular bundle.
SE induction
Two weeks after electrode implantation, each rat was transferred to a recording cage (40 × 40 × 80 cm3) and connected to a recording and stimulation system (NeuroData Digital Stimulator, Cygnus Technology Inc., Delaware Water Gap, NJ, USA) with a shielded multi-strand cable and electrical swivel (Air Precision, Le Plessis Robinson, France). A week after habituation to the new condition, rats underwent tetanic stimulation (50 Hz) of the hippocampus in the form of a succession of trains of pulses every 13 s. Each train had a duration of 10 s and consisted of biphasic pulses (pulse duration 0.5 ms, maximal intensity 500 μA). Stimulation was stopped when the rats displayed sustained forelimb clonus and salivation for minutes, which usually occurred within 1 h. However, stimulation never lasted >90 min. Behaviour was continuously monitored during electrical stimulation and several hours thereafter. Immediately after termination of the stimulation, periodic epileptiform discharges (PEDs) occurred at a frequency of 1–2 Hz and were accompanied by behavioural generalized seizures and EEG seizures SE. The total PED duration was considered as the total SE duration. In 34 rats a SE was electrically evoked, which lasted from at least 3 h, up to 13 h. Electrode implanted control rats were handled and recorded identically, but did not receive electrical stimulation.
EEG monitoring
Differential EEG signals were amplified (10×) via a FET transistor that connected the headset of the rat to a differential amplifier (20×; CyberAmp, Axon Instruments, Burlingame, CA, USA), filtered (1–60 Hz), and digitized by a computer. A seizure detection program (Harmonie, Stellate Systems, Montreal, Canada) sampled the incoming signal at a frequency of 200 Hz per channel. All EEG recordings were visually screened and seizures were confirmed by trained human observers. All rats were monitored continuously from the SE onwards, until the first spontaneous seizure appeared. Hereafter some rats were disconnected from the set-up. All rats were connected again 4 months later and continuous EEG recordings (24 h/day) were started to determine seizure frequency and duration. As previously described (Gorter et al., 2001; van Vliet et al., 2004), a stable baseline of seizure frequency is normally reached in chronic epileptic rats at this time-point, and no seizure clusters occur. Rats were monitored for at least 1 week and experiments were not started before a stable baseline was reached.
Albumin IR in rat brain
Albumin extravasation in the rat was studied by fluorescent albumin immunocytochemistry. A subset of free-floating sections that was used in Evans Blue (EB) tracer experiments (see below) were washed (2 × 10 min) in 0.05 M PBS and then incubated with anti-albumin (rabbit anti-albumin, 1:100, DakoCytomation, Glostrup, Denmark). After 24 h, the sections were washed in PBS (3 × 10 min) and incubated for 1.5 h in anti-rabbit Alexa Fluor 488 (1 : 200, Molecular Probes). Following three additional washes in PBS, sections were mounted on slides (Superfrost Plus, Menzel, Braunschweig, Germany) and coverslipped with mounting medium for fluorescence, containing 4′,6-diamidino-2-phenylindole, which labels cell nuclei (Vectashield with DAPI, Vector Laboratories, Burlingame, CA, USA). Images were acquired using a confocal-laser scanning microscope and Adobe Photoshop.
Quantification of BBB permeability
Since the detection of extravasated albumin by immunocytochemistry is not an accurate measurement to determine whether the BBB was permeable at a specific time-point, additional experiments were performed in rats. In order to quantify BBB permeability during epileptogenesis and relate changes in permeability directly to seizure activity, rats were injected with two different fluorescent tracers that do not enter the brain under normal circumstances (except for the circumventricular organs). In addition, these tracers bind to albumin, so that a comparison could be made with immunocytochemical data. Fluorescein (FSC) was used to quantify BBB permeability microscopically, while EB was used in a limited number of rats to detect BBB permeability macroscopically and to confirm the distribution of BBB permeability microscopically as detected by FSC. These tracers were intravenously (i.v.) administered via the tail vein (EB, 50 mg/kg i.v., Sigma-Aldrich, Steinheim, Germany; FSC; 100 mg/kg i.v., Merck, Darmstadt, Germany) under isoflurane anaesthesia (4 vol%). EEG recordings were discontinued during anaesthesia, which never lasted longer than several minutes. Rats were injected in the acute seizure period (1 day after SE induction; EB n = 2; FSC n = 5; and 2 days after SE; FSC n = 4), in the latent period (1 week after SE; EB n = 1; FSC n = 5) and in the chronic seizure period (4 months after SE; EB n = 2; FSC n = 7), when rats display spontaneous seizures (average seizure frequency 0.3 seizures/h). In addition, electrode implanted control rats that were not stimulated were included as well (EB n = 2; FSC n = 5). Rats were disconnected from the EEG recording set-up 2 h after tracer injection and deeply anaesthetized with pentobarbital (Nembutal, intraperitoneally (i.p.), 60 mg/kg). The animals were perfused through the ascending aorta with 100 ml of physiological salt solution, followed by 300 ml 4% paraformaldehyde/0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. The brains were post-fixed in situ overnight at 4°C, dissected and cryoprotected in 30% phosphate-buffered sucrose solution, pH 7.4. After overnight incubation at 4°C, the brains were frozen in isopentane (−30°C) and stored at −80°C until sectioning. Sagittal sections (40 μm) were cut using a sliding microtome. Sections were collected in 0.1 M phosphate buffer and processed for immunocytochemistry.
Detection of EB and FSC
To detect extravasation of the fluorescent albumin-binding dyes EB and FSC, sagittal sections were mounted on slides (Superfrost Plus, Menzel, Braunschweig, Germany) and coverslipped with mounting medium for fluorescence (Vectashield, Vector Laboratories, Burlingame, CA, USA). Tracers were detected using a confocal-laser scanning microscope (Zeiss LSM510) with appropriate filter settings (EB excitation 546 nm, emission 611 nm; FSC excitation 488 nm, emission 520 nm). Images were made using Zeiss software (Zeiss LSM Image browser) and Adobe Photoshop. A quantification of FSC sections was made for each rat using three different sections: 2.4, 3.4 and 4.6 mm lateral to bregma (Paxinos and Watson, 1998). Tracers were analysed in limbic brain regions that are thought to be involved in the generation and/or spread of seizure activity, and also in the cerebellum. The following brain regions were analysed: hippocampus (granule cell layer), entorhinal cortex (layer II/III), anterior piriform cortex (layer II/III), amygdala (basolateral amygdala nucleus), thalamus (ventral postero-medial/lateral nucleus) and cerebellum. The confocal grid (271 × 271 μm2, 15 × 15 squares) was placed on the selected brain region and the number of squares that contained a FSC signal was counted. The intensity of the FSC signal was evaluated using the histogram function in Adobe Photoshop. The average signal intensity measured in control rats (which was close to zero in all analysed regions), was used for the background correction. We constructed a ‘permeability index’ (number of squares that contained a FSC signal × FSC intensity) and all data were expressed as mean ± SEM. Statistical analysis on the permeability index was performed using ANOVA, followed by the Student's t-test. Differences with P < 0.05 were considered significant. A correlation between two ordinal variables was calculated using a Spearman's rank correlation test (P < 0.05).
Colocalization study
To confirm that EB and FSC bind to albumin and to determine whether EB and FSC colocalized with specific cell types, double labelling was performed on a subset of sections (at least two sections/rat, 2.4, 3.4 and 4.6 mm lateral to bregma) with anti-albumin (rabbit anti-albumin, 1:100, DakoCytomation, Glostrup, Denmark), the microglial marker anti-OX-42 [mouse anti-rat CD11b/c (OX-42), 1:100, PharMingen, CA, USA], the astrocytic marker anti-glial fibrillary acidic protein (mouse anti-GFAP, 1:1000, DakoCytomation, Glostrup, Denmark) and the neuronal marker anti-NeuN (mouse anti-NeuN, 1:1000, Chemicon, UK). Free-floating sections were washed (2 × 10 min) in 0.05 M PBS, followed by washing (1 × 60 min) in PBS + 0.4% bovine serum albumin (BSA). BSA was omitted in all solutions for albumin staining. Sections were then incubated in primary antibodies. After 24 h of incubation with the primary antibody, the sections were washed in PBS (3 × 10 min) and incubated for 1.5 h in Alexa Fluor 568 (FSC sections; goat anti-mouse IgG, 1:200, Molecular Probes) or Alexa Fluor 488 (EB sections; goat anti-mouse IgG Alexa, 1:200, Molecular Probes). Following three additional washes in PBS, sections were mounted on slides (Superfrost Plus, Menzel, Braunschweig, Germany) and coverslipped with mounting medium for fluorescence (Vectashield, Vector Laboratories, Burlingame, CA, USA). Images were acquired using a confocal-laser scanning microscope and Adobe Photoshop.
Fluoro-Jade B staining
To evaluate whether tracer/albumin containing cells were degenerating cells, a Fluoro-Jade B (FJB) staining was performed as described previously (Schmued and Hopkins, 2000) on a subset of sections (at least two sections/rat, 2.4, 3.4 and 4.6 mm lateral to bregma) of rats that were injected with EB. Sections were mounted on coated slides (Superfrost Plus, Menzel, Braunschweig, Germany) and dried overnight at room temperature. They were immersed in absolute alcohol for 3 min. followed by 70% ethanol for 1 min. and distilled water for 1 min. The slides were transferred to 0.06% potassium permanganate for 15 min. After rinsing with distilled water (1 min), the slides were transferred to a 0.001% polyanionic FSC derivative solution (FJB, Histo-Chem Inc., Jefferson, AR, USA) made in 0.1% acetic acid. Slides were rinsed in water, dried, immersed in xylene and coverslipped with mountant for histology (DPX, Sigma-Aldrich, Zwijndrecht, The Netherlands). Images were acquired using a confocal-laser scanning microscope and Adobe Photoshop.
Artificial opening of the BBB
To investigate whether alterations in BBB permeability could influence seizure activity, the BBB was opened with mannitol (1.5 g/kg i.v., 25% solution, once daily for 3 consecutive days) under isoflurane anaesthesia (4 vol%) in both control rats (n = 5) and in chronic epileptic rats with a stable seizure frequency (n = 8). EEG recordings were discontinued during anaesthesia, which never lasted longer than several minutes. We confirmed that this protocol resulted in BBB extravasation of FSC (data not shown) and that short isoflurane anaesthesia combined with physiological salt administration, does not influence daily seizure activity (van Vliet et al., 2006). Rats were under continuous EEG monitoring and the number of seizures and the seizure duration were evaluated before, during and after mannitol treatment. Statistical analysis was performed using the paired Student's t-test. Differences with P < 0.05 were considered significant.
Results
Albumin IR in human and rat brain
Alterations in BBB permeability, resulting in albumin extravasation, were detected using immunocytochemistry. In the human hippocampus of autopsy controls (n = 5), no albumin extravasation was observed (Fig. 1A and B). In contrast, in resected hippocampi of patients with TLE (n = 6) strong albumin IR was present in parenchyma throughout the hippocampus, next to blood vessels (Fig. 1E). Neurons and astrocytes located around these vessels were also albumin positive (Fig. 1F). Most albumin extravasation was observed in autopsy material of patients that had died during SE (n = 2). Very strong albumin IR was seen around all blood vessels within the hippocampus and cortex (Fig. 1C). In addition, many neurons and astrocytes were also highly immunoreactive (Fig. 1D). No albumin extravasation was observed in the cerebellum of these patients.
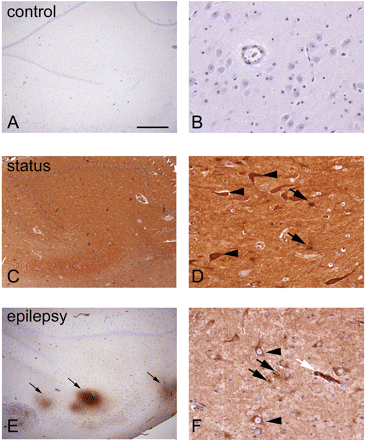
Albumin immunocytochemistry in the human hippocampus. In autopsy control tissue (A and B) no albumin staining is present. Most albumin extravasation was observed in autopsy material of patients that died during SE. Very strong albumin IR was seen around all blood vessels within the hippocampus and cortex (C and D). In addition, many neurons (arrowheads in D) and astrocytes (arrows in D) were also highly immunoreactive. In resected hippocampi from temporal lobe epilepsy patients strong albumin IR was present in parenchyma throughout the hippocampus, next to blood vessels (arrows in E). Neurons (arrowheads in F) and astrocytes (black arrows in F) were also albumin positive. The white arrow in F shows a blood vessel with albumin extravasation. Scale bar A, C and E = 800 μm, B, D and F = 75 μm.
In control rats no albumin could be detected in limbic brain regions (e.g. hippocampus, Fig. 2A). However, in the acute (1–2 days after SE) and latent phase (1 week after SE) albumin extravasation was evident in the hippocampus (Fig. 2B and C), entorhinal cortex, piriform cortex, thalamus, amygdala and olfactory bulb. In chronic epileptic rats albumin was present, but not as widespread as acutely after SE. Albumin was detected especially in the piriform cortex, but the hippocampus (Fig. 2D), entorhinal cortex, thalamus and amygdala were also immunoreactive for albumin.
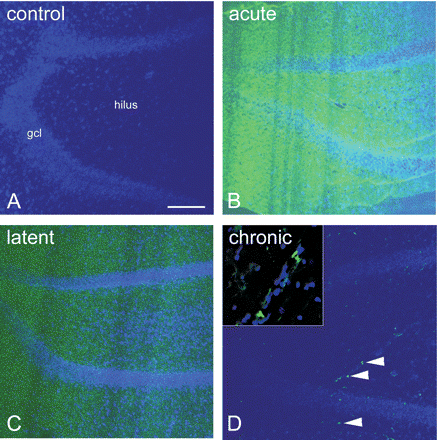
Albumin immunocytochemistry in the rat hippocampus. In control rats (A) albumin could not be detected in the dentate gyrus of the hippocampus. Sections were counterstained with DAPI (blue) to visualize cells. In the acute (B) and latent phase (C), albumin extravasation was evident throughout the dentate gyrus (green). In chronic epileptic rats (D) albumin was present in the dentate gyrus (arrowheads), but not as widespread as acutely after SE. Inset in D shows high magnification of the hilus, containing albumin particles (green). Scale bar = 100 μm, gcl = granule cell layer.
BBB permeability during epileptogenesis in rats
To assess leakage of the BBB, rats were sacrificed in the acute seizure period (1–2 days after SE), in the latent period when no seizures were observed (1 week after SE) and in the chronic epileptic period (4 months after SE), which is characterized by recurrent spontaneous seizures. The tracers EB or FSC were injected at these different time points.
Control rats
In control rats, no EB staining was observed macroscopically (Fig. 3A, D and G) or microscopically (Fig. 4A) in the analysed brain regions. The average FSC signal intensity was close to zero in all analysed regions (Fig. 6F). Since the BBB is not permeable to FSC in control rats these data were used for the background correction.
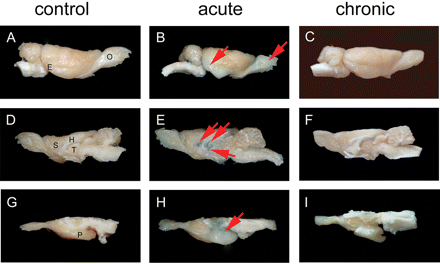
Evans Blue (EB) extravasation during epileptogenesis. In control rats, no EB staining was observed macroscopically lateral view (A); midsagittal cut (D); ventral view right half of the brain (G). In the acute seizure period, brains were oedematous and EB extravasation was observed in the hippocampus, entorhinal cortex, piriform cortex, thalamus, septum and olfactory bulb (arrows in B, E and H). No EB staining was observed in the cerebellum. In chronic epileptic rats (C, F and I) EB staining was similar to control rats. However, an increased EB staining was observed microscopically (Fig. 4). E = entorhinal cortex, O = olfactory bulb, S = septum, H = hippocampus, T = thalamus, P = piriform cortex.
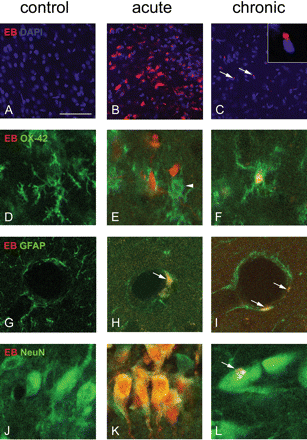
Evans Blue (EB) extravasation during epileptogenesis. In control rats, no EB staining (red) was observed microscopically (A, D, G and J). In the acute seizure period EB (red) was abundantly present (B). EB colocalized with the microglial marker OX-42 (E, arrowhead shows activated microglia migrating towards EB particle) and astrocytic marker GFAP (H) and the neuronal marker NeuN (K). In chronic epileptic rats increased EB staining was observed microscopically, mainly in layer III of the piriform cortex (C). Inset shows high power magnification. EB colocalized with reactive microglial cells (F) and astrocytes that surrounded blood vessels (I). Sparse labeling was found in neurons (L). Scale bar A–C = 100 μm, inset in C = 18 μm, D–F = 20 μm, G–I = 25 μm, J–L = 20 μm.
Acute phase
In the acute seizure phase (1 day after SE), brains were oedematous and EB extravasation was observed macroscopically in the hippocampus, entorhinal cortex, piriform cortex, thalamus, septum and olfactory bulb (Fig. 3B, E and H). In these brain regions EB was abundantly present (Fig. 4B) in neurons, as shown by colocalization with the neuronal marker NeuN (Fig. 4K). In addition, EB colocalized with the microglial marker OX-42 (Fig. 4E) and astrocytic marker GFAP (Fig. 4H), although these cells were less often observed compared with neurons that contained EB. No EB staining was observed in the cerebellum. Immunostainings with anti-albumin confirmed that EB containing cells also contained albumin (Fig. 5A–C). A large number of these cells colocalized with FJB (Fig. 5D–F), indicating degeneration. However, some neurons contained EB, but no FJB (arrowhead in Fig. 5F), which suggests that not all albumin-containing cells die.
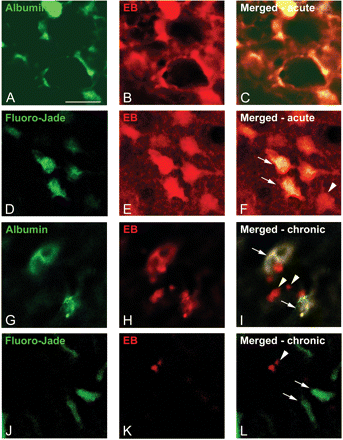
Colocalization of Albumin, Fluoro-Jade B and Evans Blue. Immunostainings with anti-albumin confirmed that EB containing cells also contained albumin, both in the acute period (1 day after SE; A–C) as well as in chronic epileptic rats (4 months after SE; arrows in I). However, not all EB particles colocalized with albumin in the chronic period (arrowheads in I). A large number of EB positive cells colocalized with Fluoro-Jade B in the acute period (arrows in F), indicating degeneration. However, some neurons contained EB, but no Fluoro-Jade B (arrowhead in F), which suggests that not all albumin containing cells die. In chronic epileptic rats most EB were found in the piriform cortex (arrowheads in L). These particles did not colocalize with Fluoro-Jade B (arrows in L). Scale bar = 20 μm.
A very large increase of FSC staining was detected in most limbic brain regions (Fig. 6G). One day after SE, the FSC permeability index increased significantly compared to controls in the parenchyma of the hippocampus, entorhinal cortex, piriform cortex, thalamus and amygdala (Fig. 6A–E). The BBB was still permeable, 2 days after SE, however, when compared with 1 day SE, the FSC permeability index decreased significantly 1.5–2 times in most analysed regions (Fig. 6). The cerebellum did not contain FSC.
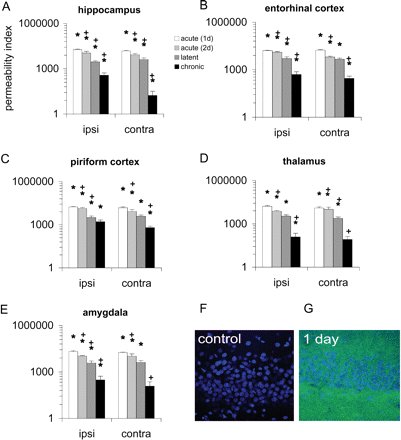
Permeability index of fluorescein (FSC) during epileptogenesis in the hippocampus (A), entorhinal cortex (B), piriform cortex (C), thalamus (D) and amygdala (E). F and G show typical examples of FSC staining (green) in the hippocampus of a control and a rat sacrificed in the acute period. Sections were counterstained with DAPI (blue) to visualize cells. While no FSC is present in the control, abundant parenchymal FSC staining is present 1 day after SE. The permeability index is expressed as mean ± SEM. In the acute seizure period, a tremendous increase of the FSC permeability index was detected in all limbic brain regions. In the latent period, the FSC permeability index decreased and was three and four times less compared with 2 days after SE in most regions, except for the entorhinal cortex in which the most intense FSC was observed (B). However, the FSC permeability index was still significantly increased compared to control rats. In chronic epileptic rats the FSC permeability index was significantly increased in the piriform cortex (C), but also in the hippocampus, entorhinal cortex, thalamus and amygdala (A, B, D and E). ‘*’ Indicates significant difference compared to control values, ANOVA followed by the Student's t-test (P < 0.05). ‘+’ Indicates significant difference compared with previous time-point (Student's t-test, P < 0.05).
Latent phase
In the latent phase (1 week after SE), EB extravasation was observed in the same brain regions as described at 1 day after SE, however to a smaller extent (data not shown). Similarly, the FSC permeability index decreased further (Fig. 6) and was 3–4 times less compared with 2 days after SE in most regions, except for the entorhinal cortex in which the most intense FSC was observed (Fig. 6B). Compared to control rats the FSC permeability index was still increased.
Chronic epileptic phase
In chronic epileptic rats (4 months after SE), EB staining was macroscopically similar to control rats (Fig. 3C, F and I). However, microscopic examination also revealed an increased EB staining, although to a much smaller extent than in the acute seizure period. Immunostainings with anti-albumin showed that EB colocalized with albumin in most cases (Fig. 5G–I). However, some EB particles did not colocalize with albumin (arrowheads in Fig. 5I). EB particles were most frequently seen in layer III of the piriform cortex (Fig. 4C), although the hippocampus, entorhinal cortex, thalamus, and amygdala also contained some EB particles. EB was mainly present in reactive microglial cells (Fig. 4F) and in astrocytes that surrounded blood vessels (Fig. 4I). Sparse labelling was found in neurons (Fig. 4L). No colocalization was found with FJB (Fig. 5J–L). The cerebellum did not contain EB.
The FSC permeability index was significantly increased compared to control rats in the piriform cortex (Fig. 6C), hippocampus, entorhinal cortex, thalamus and amygdala (Fig. 6A, B, D and E). FSC was found as ‘green particles’ with high fluorescence intensity close to cell nuclei. These particles were most evident in rats with a high seizure frequency. Similarly as has been shown for EB in Fig. 4A, FSC was mainly present in reactive microglial cells, located in layer III of the piriform cortex (data not shown). FSC was also present in astrocytes and neurons in this region, although less frequently compared with microglial colocalization.
Seizure activity and changes of BBB permeability
FSC presence in the piriform cortex (which had the highest FSC permeability index), was related with the seizure frequency (average number of seizures/hour of every chronic epileptic rat during the week before sacrifice). Figure 7A shows that a higher seizure frequency was related to a more permeable BBB (Spearman's rank, two tailed, P < 0.01, r value = 0.92). We further investigated the permeability of BBB in relation to the occurrence of the last seizure. For each rat, the time between the last seizure and sacrifice was calculated and compared with the permeability to FSC in the piriform cortex. Figure 7B shows that the time span between the last seizure and sacrifice was negatively correlated with BBB permeability (Spearman's rank, one tailed, P < 0.05, r value = −0.70). Out of the seven rats, three experienced a seizure during the presence of the tracer (shaded area in Fig. 7B). The other four rats experienced a seizure prior to tracer injection. In two of these rats FSC was also present and in the other two, in which the last seizure occurred at least 1 day before sacrifice, no FSC signal was detected.
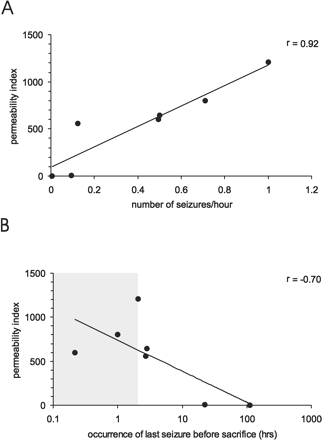
Relationship between seizure frequency and the FSC permeability index in the piriform cortex (A). A higher seizure frequency was related to a more permeable BBB (Spearman's rank, two tailed, P < 0.01, r value = 0.92). B shows the relationship between the occurrence of the last seizure and the FSC permeability index in the piriform cortex. The time span between the last seizure and sacrifice was negatively correlated with BBB permeability (Spearman's rank, one tailed, P < 0.05, r value = −0.70). The shaded area shows the presence of the tracer, which was injected 2 h before sacrifice.
To determine whether changes in BBB permeability can affect seizure activity, additional experiments were performed. In order to artificially open the BBB, both control and chronic epileptic rats were infused with mannitol. Epileptic rats that had a relatively stable daily seizure frequency (on average 4.2 ± 0.6 seizures/day, n = 8) were selected and the effect on seizure frequency was measured during and after mannitol treatment. Mannitol did not induce seizures in control rats. In contrast, the seizure frequency was significantly enhanced during the 3-day mannitol treatment in all chronic epileptic rats (compared with pre-treatment values, paired Student's t-test, P < 0.01; Fig. 8). The seizure frequency was still significantly increased in all rats during 3 days after mannitol treatment was stopped. After the first 3 days, two groups of rats could be distinguished on the basis of their seizure frequency. In the majority of rats (n = 5) the seizure frequency increased progressively over time (progressive; Fig. 8). In 3 out of 8 rats the seizure frequency returned to pre-treatment values (non-progressive). Mannitol treatment did not affect seizure duration (pre-treatment period, 60 ± 5 s; mannitol treatment, 61 ± 4 s; after mannitol treatment was stopped, 58 ± 3 s).
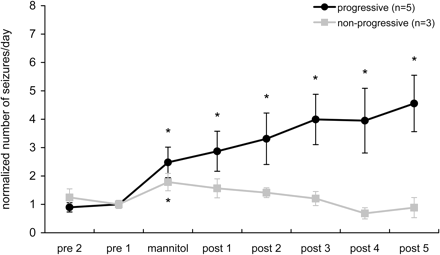
Seizure frequency in chronic epileptic rats before, during and after mannitol treatment (mean ± SEM of consecutive 3 day periods). The seizure frequency increased significantly in all chronic epileptic rats during mannitol treatment (compared with pre-treatment values, paired Student's t-test, P < 0.05). Hereafter, two groups of rats could be distinguished on the basis of their seizure frequency. In the majority of rats (n = 5) the seizure frequency increased progressively over time (progressive). In 3 out of 8 rats the seizure frequency returned to pre-treatment values (non-progressive). ‘*’ Indicates significant difference compared with pre-treatment values (paired Student's t-test P < 0.05).
Discussion
The main new findings of our study are: (i) opening of the BBB is likely to occur after a single seizure in the chronic epileptic phase; and (ii) transient opening of the BBB by hyperosmotic treatment could aggravate epileptic seizures, often in a persistent manner.
The BBB was disrupted in epileptic rats and humans, both shortly after SE as well as in the chronic epileptic phase. In rats, we showed that the BBB is not only open during a seizure but remains open for at least 1 h thereafter. Nevertheless, rats that just experienced a seizure had a more permeable BBB, indicating that the BBB is even more open during a spontaneous seizure. Changes in BBB integrity may be caused by increased local blood pressure that occurs during seizures (Nitsch and Klatzo, 1983; Oztas and Kaya, 1991), free radical formation (Oztas et al., 2001), inflammatory responses such as leucocyte recruitment, cytokine and interleukin production (Patel, 2004; Vezzani and Granata, 2005; Gorter et al., 2006) and/or loss of tight junction molecules (Ballabh et al., 2004).
One of the consequences of increased BBB permeability is the accumulation of serum proteins that enter the brain, which may contribute to increased excitability. It has been shown recently that epileptiform activity can be induced by direct cortical application of albumin-containing solution in rats, suggesting that serum proteins play a role in the pathogenesis of focal epilepsies (Seiffert et al., 2004). Interestingly, albumin was found specifically in regions with more EEG spiking activity in human (Cornford et al., 1998) and induced calcium waves in rat cortical astrocytes (Nadal et al., 1997). We showed a positive correlation between BBB permeability and the occurrence of spontaneous seizures in chronic epileptic rats. The increased BBB permeability was mainly observed in the piriform cortex, which is considered to be highly epileptogenic (Loscher and Ebert, 1996; Demir et al., 1999; Ebert et al., 2000). However, several other limbic regions also showed BBB leakage, although to lesser extent. This positive correlation and the finding that artificial opening of the BBB by mannitol increased seizure frequency in chronic epileptic rats add up to the increasing evidence that BBB leakage could contribute to the increased excitability or maintenance of the epileptic condition. The fact that the rats do not experience seizures during the latent period when the BBB leakage is many times higher that during the chronic epileptic phase might be due to other factors such as transient decreased expression of genes related to synaptic transmission which has been observed during the latent period (Gorter et al., 2006). Moreover, leakage of the BBB early in the process of epileptogenesis may not directly cause seizure activity as suggested by the study of Seiffert et al. (2004), in which increased excitability was not observed until 4–7 days after BBB disruption.
In contrast, acute opening of the BBB in chronic epileptic rats increased the seizure frequency in all rats. In the majority of rats (62%) this led to a permanent and progressive increase in the number of seizures later on. Interestingly, one of the complications of BBB opening by mannitol in humans to treat brain tumours is the occurrence of seizures in 4–50% of the patients (Neuwelt et al., 1983, 1986; Roman-Goldstein et al., 1994). In rats, increased BBB opening did not necessarily lead to a persistent seizure progression, since in a subset of rats (38%) the number of spontaneous seizures returned to pre-mannitol treatment levels. The specific mechanisms that may induce increased excitability after BBB disruption are not known, but it has been hypothesized that slow processes regulated at the transcriptional–translational level (triggered by the increased concentration of proteins in the brain extracellular fluid), activation of astrocytes, impaired potassium buffering and the development of calcium waves in astrocytes may be a few of the causes (Nadal et al., 1997; Seiffert et al., 2004).
Besides hyperexcitability, serum proteins may be involved in neurodegeneration. Albumin was mainly found in neurons in epileptic tissue of human as well as in the rat shortly after SE, when cell death occurs (Gorter et al., 2003). Many cells colocalized with FJB, indicating that these cells were degenerating cells. Similarly, neuronal accumulation of serum proteins after intracerebral haemorrhage was associated with cytochrome c release, DNA fragmentation, and cell death (Matz et al., 2001). Neuronal accumulation of albumin has also been shown after induced seizures (Nitsch and Klatzo, 1983; Sokrab et al., 1989) and ischemia (Loberg et al., 1993, 1994).
In addition to the presence of albumin in neurons, astrocytes also contained albumin, which is in agreement with previous rat (Lassmann et al., 1984) and human studies (Mihaly and Bozoky, 1984). The role of albumin-containing astrocytes is unclear, but it has been hypothesized that astrocytes may have a protective function by clearing extravasated albumin from the extracellular space (Mihaly and Bozoky, 1984).
Albumin deposits were also found in microglial cells in the rat brain. Shortly after SE, they were abundantly present in many limbic brain regions, while in the chronic epileptic phase they were mainly restricted to layer II and III of the piriform cortex. These cells are thought to be involved in trapping serum-derived foreign substances that enter the brain (Xu and Ling, 1994); especially in regions where BBB integrity is affected. Interestingly, in those limbic regions we previously reported increased expression of ferritin, an iron-storage protein that may be induced by local extravasation of blood and release of iron from haemoglobin-containing blood cells (Gorter et al., 2005).
In conclusion, this study shows dynamic changes of BBB permeability during epileptogenesis. The long-lasting increased permeability of the BBB was present in various limbic regions and correlated to seizure activity suggesting that it may contribute to increased excitability in the epileptogenic foci that can lead to progression of epilepsy. Given that our study suggests that a compromised BBB could contribute to seizure development and progression of epilepsy, the BBB may represent an adequate target to intervene with epileptogenesis. In this respect, agents that can alter BBB permeability are candidates for strategies in order to control the progression of epilepsy.
We thank Prof. Dr F. H. Lopes da Silva and Prof. Dr W. J. Wadman for critically reading the manuscript. Part of this work was supported by the Epilepsy Institute of The Netherlands. We would like to thank Dr W. G. M. Spliet and Prof. Dr D. Troost (neuropathologists; Department of (Neuro)Pathology of University Medical Center Utrecht and University of Amsterdam) for the collaboration in the collection of human material. E.A. and J.A.G. are supported by the National Epilepsy Fund—‘Power of the Small’ and Hersenstichting Nederland [NEF 02-10 and NEF 05-11 (E.A.) and 03-03 (J.G.)].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}