




Abstract
The relationship between severe myoclonic epilepsy of infancy (SMEI or Dravet syndrome) and the related syndrome SMEI-borderland (SMEB) with mutations in the sodium channel alpha 1 subunit gene SCN1A is well established. To explore the phenotypic variability associated with SCN1A mutations, 188 patients with a range of epileptic encephalopathies were examined for SCN1A sequence variations by denaturing high performance liquid chromatography and sequencing. All patients had seizure onset within the first 2 years of life. A higher proportion of mutations were identified in patients with SMEI (52/66; 79%) compared to patients with SMEB (25/36; 69%). By studying a broader spectrum of infantile epileptic encephalopathies, we identified mutations in other syndromes including cryptogenic generalized epilepsy (24%) and cryptogenic focal epilepsy (22%). Within the latter group, a distinctive subgroup designated as severe infantile multifocal epilepsy had SCN1A mutations in three of five cases. This phenotype is characterized by early onset multifocal seizures and later cognitive decline. Knowledge of an expanded spectrum of epileptic encephalopathies associated with SCN1A mutations allows earlier diagnostic confirmation for children with these devastating disorders.
Introduction
SCN1A, the gene encoding the sodium channel alpha 1 subunit, has emerged as the most important of the epilepsy genes currently known (Mulley et al., 2005). SCN1A mutations underlie more than 70% of patients with the epileptic encephalopathy severe myoclonic epilepsy of infancy (SMEI or Dravet syndrome) (Dravet et al., 1982; Claes et al., 2001; Mulley et al., 2005). More than 170 documented mutations are associated with SMEI and the related syndrome of borderland SMEI, known as SMEB. Truncation mutations account for nearly 50% of mutations found in SMEI, with the remainder comprising missense, splice site and deletion mutations (Mulley et al., 2005). These mutations affect many domains of the gene with suggested clustering of missense mutations occurring in the N- and C-termini and the S5–S6 pore-forming regions of the protein (Kanai et al., 2004). Recently intragenic and whole gene deletions have been identified in a few cases of SMEI without truncation, missense or splice-site mutations (Madia et al., 2006; Mulley et al., 2006; Suls et al., 2006).
Approximately 95% of SCN1A mutations in SMEI patients arise de novo. The remaining cases have familial mutations with milder phenotypes in other family members often consistent with the generalized epilepsy with febrile seizures plus (GEFS+) spectrum (Scheffer and Berkovic, 1997; Escayg et al., 2000; Singh et al., 2001; Fujiwara et al., 2003; Nabbout et al., 2003a; Scheffer, 2003; Kimura et al., 2005). Recently, germline and somatic SCN1A mutational mosaicism has been reported in unaffected parents (or parents mildly affected with febrile seizures) where their children have SMEI or SMEB (Depienne et al., 2006; Gennaro et al., 2006; Marini et al., 2006; Morimoto et al., 2006).
SMEI or Dravet syndrome is a distinctive syndrome with seizure onset in the first year of life, typically beginning with prolonged febrile hemiclonic seizures or generalized tonic–clonic seizures (Dravet, 1978; Dravet et al., 1982, 2005). The disorder evolves with other seizure types such as myoclonic, focal, absence and atonic seizures developing between 1 and 4 years of age. Development is normal in the first year of life followed by developmental slowing and regression. Pyramidal signs and ataxia may evolve. Cognitive outcome is usually poor and seizures remain refractory for those who survive to adulthood (Jansen et al., 2006).
The phenotypic spectrum of patients with SCN1A mutations has been extended beyond SMEI. The related syndrome SMEB (Ohmori et al., 2003; Fukuma et al., 2004) refers to children who lack several of the key features of SMEI such as myoclonic seizures or generalized spike-wave activity (Sugama et al., 1987; Dravet et al., 2005). In two studies, 26% (7/27) and 88% (15/17) of SMEB patients were found to have SCN1A mutations respectively (Ohmori et al., 2003; Fukuma et al., 2004). As with SMEI, these mutations are spread throughout the gene with a mixture of types of mutation including truncation, missense and splice-site changes (Mulley et al., 2005). A subgroup of SMEB has been variously described as intractable childhood epilepsy with generalized tonic clonic seizures (ICEGTC, originally called high voltage slow waves grand mal by Japanese authors) or Severe idiopathic generalized epilepsy of infancy with generalized tonic–clonic seizures. These infants have generalized tonic–clonic seizures beginning in the first year of life without the evolution of other seizure types and they follow a similarly unfavourable developmental course to children with SMEI (Fujiwara et al., 1992; Kanazawa, 1992, 2001; Sugama et al., 1987; Doose et al., 1998). In one series, 7/10 ICEGTC patients had missense mutations in SCN1A (Fujiwara et al., 2003); truncation, missense and splice-site mutations were reported in 3/18 patients described as severe idiopathic generalized epilepsy of infancy (Ebach et al., 2005). We reported the only case so far of West syndrome with an SCN1A mutation (Wallace et al., 2003).
Given the overlapping yet heterogeneous clinical features of these epilepsy syndromes, we postulated that SCN1A mutations may be associated with other phenotypes. Here we studied unselected patients with severe epileptic encephalopathies (including SMEI) with onset primarily during the first year of life.
Material and methods
Clinical methods
Patients with epileptic encephalopathies of unknown cause were referred by paediatric neurologists and neurologists from Australia and around the world. Epileptic encephalopathies are defined as disorders in which there is a temporal relationship between deterioration in cognitive, sensory and motor function and epileptic activity comprising frequent seizures and/or extremely frequent ‘interictal’ paroxysmal activity (Nabbout and Dulac, 2003). Cases were only included where magnetic resonance imaging was normal or showed non-specific features without a definite aetiology. A subset of 14 patients, included in this study, with so-called ‘vaccine encephalopathy’ has been published previously (Berkovic et al., 2006).
Electroclinical data were obtained on all patients with specific emphasis on early seizure history including age of onset, occurrence of status epilepticus, presence of fever sensitivity, clinical photic sensitivity and evolution of other seizure types. A detailed early developmental history was obtained with attention to acquisition of early milestones, timing of plateau or regression of development and current functioning. Other important details included general and neurological examination, family history of seizure disorders and results of EEG, video-EEG monitoring and neuroimaging studies. Results of other available investigations such as chromosomal analysis were also obtained.
SMEI was defined according to the following criteria: onset in the first year of life of convulsive seizures which were hemiclonic or generalized; myoclonic seizures; other seizure types which could include focal seizures, absence seizures, atonic seizures, tonic seizures; normal development in the first year of life with subsequent slowing including plateauing or regression; generalized spike-wave activity and either normal MRI or non-specific findings.
SMEB was divided into subgroups based on the absence of specific features that are regarded as required for the diagnosis of SMEI. SMEB-M referred to patients who did not have myoclonic seizures but otherwise satisfied SMEI criteria. SMEB-SW defined patients who had all the SMEI criteria but had never had generalized spike-wave activity documented on EEG. SMEB-O referred to patients who had more than one feature that was not in keeping with SMEI; examples include absence of generalized spike-wave activity recorded on EEG, a normal developmental outcome and absence of myoclonic seizures. SMEB included cases with ICEGTC where they followed the same course but only had convulsive seizures.
Cryptogenic generalized epilepsy (CGE) denoted individuals who have multiple seizure types, generalized sharp and slow activity and intellectual disability with no known aetiology. Lennox–Gastaut syndrome (LGS) defined patients with tonic seizures and slow generalized spike-wave activity and abnormal development (>Commission on Classification and Terminology of the International League Against Epilepsy, 1989; Beaumanoir and Blume, 2005). Myoclonic–astatic epilepsy (MAE) referred to individuals with myoclonic–astatic seizures and other generalized seizure types with generalized spike-wave activity and variable developmental outcome (Doose et al., 1970; Guerrini et al., 2005).
A further subgroup was called cryptogenic focal epilepsy where an individual had focal seizures and uni- or multifocal EEG epileptiform patterns. These individuals showed a variable degree of intellectual disability and usually had normal neuroimaging. Several individuals were included with abnormal neuroimaging that did not account for the clinical presentation such as hydrocephalus, bilateral periventricular leucomalacia, etc.
Other syndromes were defined according to the ILAE classification (Commission on Classification and Terminology of the International League Against Epilepsy, 1989). Patients were called ‘unclassified’ if we had insufficient evidence to make a syndrome diagnosis or the patient did not fit into a recognized syndrome despite detailed evaluation.
The Austin Health Human Research Ethics Committee approved this study. Informed consent was obtained from the parents or guardians of minors and from adult subjects of normal intellect. In the case of adults with intellectual disability, legal consent was obtained from the appropriate government authority or legal guardian.
Molecular analysis
Molecular analysis was carried out on genomic DNA extracted from venous blood. All 26 exons of SCN1A were amplified by polymerase chain reaction (PCR) using flanking intronic primers and standard PCR conditions (primers available upon request). PCR fragments were heat denatured at 95°C for 4 min and slowly cooled to room temperature to form heteroduplex products which were analysed by denaturing high performance liquid chromatography (dHPLC) on the Transgenomic WAVE 3500HT instrument (dHPLC conditions available upon request). Amplicons showing altered dHPLC chromatogram patterns were sequenced in both directions from independent PCR products, on an ABI 3700 sequencer. The final subset of patients (43) was screened by direct sequencing of PCR products (without prior dHPLC screening) by Athena Diagnostics under diagnostic conditions. The numbering for each mutation is taken from the start codon ATG of the full-length SCN1A isoform sequence (Genbank accession number AB093548). In cases where an SCN1A mutation was detected, the appropriate amplicon from parental DNA (where available) was tested by DNA sequencing to distinguish between de novo and familial variants. Mutations or rare variants were distinguished from coding single nucleotide polymorphisms which have previously been reported (Escayg et al., 2001).
Results
Clinical diagnoses
One hundred and eighty-eight patients were recruited from Australia (110), Canada (27), United Kingdom (23), New Zealand (20), Israel (4), USA (3) and Denmark (1) with seizure onset in the first 2 years of life. These included 14 cases who were negative for SCN1A mutations on single-stranded conformation analysis in our previous study (Wallace et al., 2003); the eight positive cases and two, who were negative on sequencing, are not included in the data presented here.
Our total cohort contained 66 with SMEI, 36 with SMEB including the various subcategories, 25 with cryptogenic generalized epilepsy, 18 with cryptogenic focal epilepsy, 10 with MAE and 12 with LGS. The remaining cases had a range of other syndromes or were unable to be classified (Table 1).
SCN1A mutations in patients with epileptic encephalopathies
| Total | SCN1A mutation | |
|---|---|---|
| SMEI | 66 | 52 |
| SMEB | 36 | 25 |
| SMEB-O | 16 | 10 |
| SMEB-SW | 14 | 11 |
| SMEB-M | 4 | 3 |
| ICEGTC | 2 | 1 |
| Cryptogenic generalized epilepsy | 25 | 6 |
| Cryptogenic focal epilepsy | 18 | 4 |
| Myoclonic–astatic epilepsy | 10 | 2 |
| Lennox–Gastaut syndrome | 12 | 1 |
| West syndrome | 5 | |
| Idiopathic spasms | 1 | |
| Early myoclonic encephalopathy | 1 | |
| Progressive myoclonic epilepsy | 1 | |
| Alternating hemiplegia of childhood | 1 | |
| Unclassified | 12 | |
| Total | 188 | 90 |
| Total | SCN1A mutation | |
|---|---|---|
| SMEI | 66 | 52 |
| SMEB | 36 | 25 |
| SMEB-O | 16 | 10 |
| SMEB-SW | 14 | 11 |
| SMEB-M | 4 | 3 |
| ICEGTC | 2 | 1 |
| Cryptogenic generalized epilepsy | 25 | 6 |
| Cryptogenic focal epilepsy | 18 | 4 |
| Myoclonic–astatic epilepsy | 10 | 2 |
| Lennox–Gastaut syndrome | 12 | 1 |
| West syndrome | 5 | |
| Idiopathic spasms | 1 | |
| Early myoclonic encephalopathy | 1 | |
| Progressive myoclonic epilepsy | 1 | |
| Alternating hemiplegia of childhood | 1 | |
| Unclassified | 12 | |
| Total | 188 | 90 |
SMEI, severe myoclonic epilepsy of infancy; SMEB-SW, SMEI borderland without generalized spike wave; SMEB-M, SMEI borderland without myoclonic seizures; SMEB-O, SMEI borderland lacking more than one feature of SMEI; ICEGTC, intractable childhood epilepsy with generalized tonic–clonic seizures.
SCN1A mutations in patients with epileptic encephalopathies
| Total | SCN1A mutation | |
|---|---|---|
| SMEI | 66 | 52 |
| SMEB | 36 | 25 |
| SMEB-O | 16 | 10 |
| SMEB-SW | 14 | 11 |
| SMEB-M | 4 | 3 |
| ICEGTC | 2 | 1 |
| Cryptogenic generalized epilepsy | 25 | 6 |
| Cryptogenic focal epilepsy | 18 | 4 |
| Myoclonic–astatic epilepsy | 10 | 2 |
| Lennox–Gastaut syndrome | 12 | 1 |
| West syndrome | 5 | |
| Idiopathic spasms | 1 | |
| Early myoclonic encephalopathy | 1 | |
| Progressive myoclonic epilepsy | 1 | |
| Alternating hemiplegia of childhood | 1 | |
| Unclassified | 12 | |
| Total | 188 | 90 |
| Total | SCN1A mutation | |
|---|---|---|
| SMEI | 66 | 52 |
| SMEB | 36 | 25 |
| SMEB-O | 16 | 10 |
| SMEB-SW | 14 | 11 |
| SMEB-M | 4 | 3 |
| ICEGTC | 2 | 1 |
| Cryptogenic generalized epilepsy | 25 | 6 |
| Cryptogenic focal epilepsy | 18 | 4 |
| Myoclonic–astatic epilepsy | 10 | 2 |
| Lennox–Gastaut syndrome | 12 | 1 |
| West syndrome | 5 | |
| Idiopathic spasms | 1 | |
| Early myoclonic encephalopathy | 1 | |
| Progressive myoclonic epilepsy | 1 | |
| Alternating hemiplegia of childhood | 1 | |
| Unclassified | 12 | |
| Total | 188 | 90 |
SMEI, severe myoclonic epilepsy of infancy; SMEB-SW, SMEI borderland without generalized spike wave; SMEB-M, SMEI borderland without myoclonic seizures; SMEB-O, SMEI borderland lacking more than one feature of SMEI; ICEGTC, intractable childhood epilepsy with generalized tonic–clonic seizures.
Molecular analysis
Of the 188 patients examined, 90 (48%) had SCN1A mutations. Ninety-four sequence variants were identified in the 90 mutation positive patients as four children each had two changes. In each child, the putative pathogenic variant was distinguished from the likely non-pathogenic variant; the latter was not included in further analyses (see later and Supplementary Table). The majority of mutations are novel (72/90, 80%), reinforcing the mutational heterogeneity characteristic of SCN1A. Of the 90 cases, DNA was available from 76 sets of parents and 73/76 (96%) were de novo mutations.
Amino acid alignments of the missense mutations show that they affect conserved domains of the protein in other human alpha channels (SCN2A, SCN3A and SCN8A), chimpanzee, rat, mouse, Fugu and Drosophila consistent with their interpretation as pathogenic mutations (Supplementary Fig. S1). Moreover, the probability that these missense mutations are pathogenic mutations is supported by their de novo origin (in 34/37 cases where parents have been examined) and previously published observations in SMEI.
SMEI
Fifty-two of the 66 (79%) of patients with SMEI had SCN1A mutations (Table 1). Forty-four percent (23/52) of the SMEI-related mutations were non-sense or frameshift mutations resulting in protein truncation, 39% (20/52) were missense mutations and the remaining 17% (9/52) were intronic splice donor or splice acceptor site changes. These mutations were spread throughout the gene with the majority of missense mutations (14/20, 70%) localized to the transmembrane regions of the protein, in particular the S5–S6 loop of domain II that forms part of the ion channel pore (Supplementary Table, Fig. 1A). In contrast, 57% (13/23) of truncation mutations were positioned in the intracellular loops of the protein (Supplementary Table, Fig. 1A).
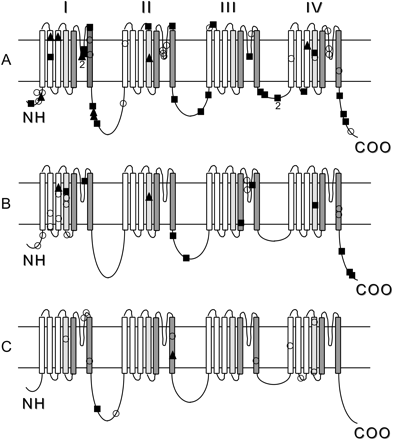
Schematic representation of mutations in SCN1A in patients with (A) SMEI, (B) SMEB and (C) other phenotypes. Refer to Supplementary Table for details. The SCN1A protein consists of four domains designated I–IV, each contains six transmembrane segments designated S1–S6. ▪ = truncation, ○ = missense, ▴ = splice-site mutations.
Parental DNA was available for testing for 42/52 SMEI patients who were mutation positive. Analysis of the DNA from these parents confirmed that all 42 mutations were de novo.
There were four patients with two sequence variants that posed challenges in clinico-molecular interpretation (Supplementary Table). Patient 2 had two SCN1A sequence variants: one was a de novo missense change (Y84C) affecting a highly conserved amino acid site (Supplementary Fig. S1) and the second was a splice acceptor site change found to have a maternal origin. The mother was unaffected; the maternal grandfather had a history of convulsions until 7 years but was negative for the splice acceptor site change. There was no further seizure history within this family suggesting that the change within the splice site was probably a benign variant. Therefore this variant was not considered in the determination of mutation frequencies.
Patients 6, 32 and 38 also had two sequence variants detected but parental DNA was unavailable in order to ascertain which variant was de novo and thus the likely pathogenic mutation (Supplementary Table). Patient 6 had two intronic mutations detected, both potentially pathogenic. In the absence of parental DNA we can only assume that one is likely to be pathogenic. The intron IVS3-13T→A change was chosen as the most likely variant to affect splicing since it is within the consensus C/T run in the splice acceptor site. Patient 32 had both a missense (E1238D) and an intronic donor splice site mutation. Since the missense change affected a highly conserved amino acid site (Supplementary Fig. S1), this was considered to be the true mutation. Patient 38 had a truncation mutation (R1525X) also seen in Patient 39 with SMEI and in a previous study (Supplementary Table) (Kearney et al., 2006). Patient 38 also had a missense change not as highly conserved as most missense mutations (Supplementary Fig. S1) so the truncation mutation was considered the likely pathogenic mutation. The second variant found in each case has not been included in the mutational analyses.
Simultaneous double mutation in the same patient is a theoretical possibility, as is a de novo mutation adversely interacting with a pre-existing rare variant. However, in the absence of definitive evidence from other SMEI cases and absence of parental DNA to establish de novo origin, the most parsimonious explanation is that of a single mutational event unless proven otherwise.
SMEI—borderland
SCN1A mutations were identified in 25/36 (69%) patients with SMEB including all subcategories (Table 1). Over half of these changes were missense mutations (13/25) with 40% (10/25) being truncation mutations; the remaining two were splice-site mutations. The mutations were spread throughout the gene with the majority (18/25, 72%) localized to the transmembrane domain regions. Missense mutations were clustered in the S2–S4 transmembrane segments of domain I (Fig. 1B).
Analysis of parental DNA from 22/25 patients with mutations confirmed 95% (21/22) were de novo. Patient 63 had a paternally inherited mutation (A239T). The proband's father had febrile seizures plus (Scheffer and Berkovic, 1997) and the paternal grandmother had unclassified seizures. Both individuals were found to carry the A239T change, which when taken together with the amino acid conservation of this residue (Supplementary Fig. S1), reinforces the status of this variant as a true pathogenic mutation of SCN1A. The family had a bilineal family history of seizures as the proband's mother had febrile seizures and did not carry the SCN1A mutation (Supplementary Fig. S2).
Cryptogenic generalized epilepsy
Of the 25 patients with cryptogenic generalized epilepsy, six (24%) had mutations. None of the mutations have been previously reported, however the T226M in Patient 78 was also seen in Patient 61 within this cohort with SMEB-O (Supplementary Table). Four mutations arose de novo; parental DNA was unavailable for one patient and for Patient 82 the mutation (M973V) was found in her unaffected father. There was no family history of seizures but the amino acid conservation at this site is reasonably strong (Supplementary Fig. S1) providing circumstantial evidence that it is a true mutation. If so, then it must be non-penetrant in the father or else function as a susceptibility allele acting in tandem with other unidentified susceptibility genes responsible for the phenotype in the proband.
The six cases with mutations had heterogeneous phenotypes with onset between 1.5 and 12 months (Table 2). Two had a phenotype with onset in the first 2 months of life and abnormal early development but other features were similar to SMEI. Patient 80 fixed and followed and smiled by 6 weeks when seizures began. Development slowed from 6 weeks: he sat late, walked at 18 months and development stagnated from 2 years. He died at 13 years. Patient 78 had seizure onset at 8 weeks, smiled at 3 months, never sat or acquired words.
Clinical features of SCN1A mutation positive patients with diagnosis other than SMEI and SMEB
| Patient | Age at study (years) | Seizure onset (months) | Seizure types | Intellect | Neurological signs | Epilepsy classification | SCN1A mutation | Inheritance |
|---|---|---|---|---|---|---|---|---|
| 78 | 5 | 2 | GTCS, H, MJ, F, NCS | Severe ID | Increased tone, later generalized hypotonia | CGE | T226M | De novo |
| 79 | 23 | 5.5 | FS, GTCS | Mild-moderate ID | None | CGE | A395P | De novo |
| 80 | 14 | 1.5 | GTCS, H, At, MJ, F, SE | ID | None | CGE | V422E | De novo |
| 81 | 14 | 12 | FS, GTCS, aAb, MJ, SE | ID | Ataxia, intermittent movement disorder | CGE | S626G | ND |
| 82 | 35 | 9 | FS, GTCS, MJ, F-SG | Low average | None | CGE | M973V | Paternal |
| 83 | 3 | 6 | FS, GTCS, MJ | Normal | None | CGE | IVS15+1G→T | De novo |
| 84 | 16 | 7 | FS, GTCS, MJ, F, NCS | Mild ID | Mild generalized spasticity | CFE (SIMFE) | F575fsX622 | De novo |
| 85 | 5 | 4.5 | F, H, SE | ID | None | CFE (SIMFE) | F1543S | Maternal |
| 86 | 20 | 5 | GTCS, MJ, F, T | Moderate ID | Ataxia, mild left hemiparesis | CFE (SIMFE) | R1596C | De novo |
| 87 | 5 | 18 | FS, F-SG, SE | Normal | None | CFE | R1657H | De novo |
| 88 | 21 | 0.75 | IS, At, aAb, T, SE, NCS | ID | Mild right hemiparesis | LGS | R1636Q | De novo |
| 89 | 11 | 4 | FS, GTCS, MJ, At, T, H | ID | None | MAE | R393C | De novo |
| 90 | 12 | 13 | FS, F, MA, MJ | Moderate ID | None | MAE | G1480V | De novo |
| Patient | Age at study (years) | Seizure onset (months) | Seizure types | Intellect | Neurological signs | Epilepsy classification | SCN1A mutation | Inheritance |
|---|---|---|---|---|---|---|---|---|
| 78 | 5 | 2 | GTCS, H, MJ, F, NCS | Severe ID | Increased tone, later generalized hypotonia | CGE | T226M | De novo |
| 79 | 23 | 5.5 | FS, GTCS | Mild-moderate ID | None | CGE | A395P | De novo |
| 80 | 14 | 1.5 | GTCS, H, At, MJ, F, SE | ID | None | CGE | V422E | De novo |
| 81 | 14 | 12 | FS, GTCS, aAb, MJ, SE | ID | Ataxia, intermittent movement disorder | CGE | S626G | ND |
| 82 | 35 | 9 | FS, GTCS, MJ, F-SG | Low average | None | CGE | M973V | Paternal |
| 83 | 3 | 6 | FS, GTCS, MJ | Normal | None | CGE | IVS15+1G→T | De novo |
| 84 | 16 | 7 | FS, GTCS, MJ, F, NCS | Mild ID | Mild generalized spasticity | CFE (SIMFE) | F575fsX622 | De novo |
| 85 | 5 | 4.5 | F, H, SE | ID | None | CFE (SIMFE) | F1543S | Maternal |
| 86 | 20 | 5 | GTCS, MJ, F, T | Moderate ID | Ataxia, mild left hemiparesis | CFE (SIMFE) | R1596C | De novo |
| 87 | 5 | 18 | FS, F-SG, SE | Normal | None | CFE | R1657H | De novo |
| 88 | 21 | 0.75 | IS, At, aAb, T, SE, NCS | ID | Mild right hemiparesis | LGS | R1636Q | De novo |
| 89 | 11 | 4 | FS, GTCS, MJ, At, T, H | ID | None | MAE | R393C | De novo |
| 90 | 12 | 13 | FS, F, MA, MJ | Moderate ID | None | MAE | G1480V | De novo |
FS, febrile seizures; aAb, atypical absence seizures; At, atonic seizures; F, focal seizures (not hemiclonic/unilateral); GTCS, generalized tonic–clonic seizures; H, hemiclonic; IS, infantile spasms; MA, myoclonic–astatic; MJ, myoclonic jerks; NCS, non-convulsive status epilepticus; SE, status epilepticus; SG, secondary generalization; T, tonic seizures; CGE, cryptogenic generalized epilepsy; CFE, cryptogenic focal epilepsy; LGS, Lennox–Gastaut syndrome; MAE, myoclonic–astatic epilepsy; SIMFE, severe infantile multifocal epilepsy; ID, intellectual disability; ND, not done.
Clinical features of SCN1A mutation positive patients with diagnosis other than SMEI and SMEB
| Patient | Age at study (years) | Seizure onset (months) | Seizure types | Intellect | Neurological signs | Epilepsy classification | SCN1A mutation | Inheritance |
|---|---|---|---|---|---|---|---|---|
| 78 | 5 | 2 | GTCS, H, MJ, F, NCS | Severe ID | Increased tone, later generalized hypotonia | CGE | T226M | De novo |
| 79 | 23 | 5.5 | FS, GTCS | Mild-moderate ID | None | CGE | A395P | De novo |
| 80 | 14 | 1.5 | GTCS, H, At, MJ, F, SE | ID | None | CGE | V422E | De novo |
| 81 | 14 | 12 | FS, GTCS, aAb, MJ, SE | ID | Ataxia, intermittent movement disorder | CGE | S626G | ND |
| 82 | 35 | 9 | FS, GTCS, MJ, F-SG | Low average | None | CGE | M973V | Paternal |
| 83 | 3 | 6 | FS, GTCS, MJ | Normal | None | CGE | IVS15+1G→T | De novo |
| 84 | 16 | 7 | FS, GTCS, MJ, F, NCS | Mild ID | Mild generalized spasticity | CFE (SIMFE) | F575fsX622 | De novo |
| 85 | 5 | 4.5 | F, H, SE | ID | None | CFE (SIMFE) | F1543S | Maternal |
| 86 | 20 | 5 | GTCS, MJ, F, T | Moderate ID | Ataxia, mild left hemiparesis | CFE (SIMFE) | R1596C | De novo |
| 87 | 5 | 18 | FS, F-SG, SE | Normal | None | CFE | R1657H | De novo |
| 88 | 21 | 0.75 | IS, At, aAb, T, SE, NCS | ID | Mild right hemiparesis | LGS | R1636Q | De novo |
| 89 | 11 | 4 | FS, GTCS, MJ, At, T, H | ID | None | MAE | R393C | De novo |
| 90 | 12 | 13 | FS, F, MA, MJ | Moderate ID | None | MAE | G1480V | De novo |
| Patient | Age at study (years) | Seizure onset (months) | Seizure types | Intellect | Neurological signs | Epilepsy classification | SCN1A mutation | Inheritance |
|---|---|---|---|---|---|---|---|---|
| 78 | 5 | 2 | GTCS, H, MJ, F, NCS | Severe ID | Increased tone, later generalized hypotonia | CGE | T226M | De novo |
| 79 | 23 | 5.5 | FS, GTCS | Mild-moderate ID | None | CGE | A395P | De novo |
| 80 | 14 | 1.5 | GTCS, H, At, MJ, F, SE | ID | None | CGE | V422E | De novo |
| 81 | 14 | 12 | FS, GTCS, aAb, MJ, SE | ID | Ataxia, intermittent movement disorder | CGE | S626G | ND |
| 82 | 35 | 9 | FS, GTCS, MJ, F-SG | Low average | None | CGE | M973V | Paternal |
| 83 | 3 | 6 | FS, GTCS, MJ | Normal | None | CGE | IVS15+1G→T | De novo |
| 84 | 16 | 7 | FS, GTCS, MJ, F, NCS | Mild ID | Mild generalized spasticity | CFE (SIMFE) | F575fsX622 | De novo |
| 85 | 5 | 4.5 | F, H, SE | ID | None | CFE (SIMFE) | F1543S | Maternal |
| 86 | 20 | 5 | GTCS, MJ, F, T | Moderate ID | Ataxia, mild left hemiparesis | CFE (SIMFE) | R1596C | De novo |
| 87 | 5 | 18 | FS, F-SG, SE | Normal | None | CFE | R1657H | De novo |
| 88 | 21 | 0.75 | IS, At, aAb, T, SE, NCS | ID | Mild right hemiparesis | LGS | R1636Q | De novo |
| 89 | 11 | 4 | FS, GTCS, MJ, At, T, H | ID | None | MAE | R393C | De novo |
| 90 | 12 | 13 | FS, F, MA, MJ | Moderate ID | None | MAE | G1480V | De novo |
FS, febrile seizures; aAb, atypical absence seizures; At, atonic seizures; F, focal seizures (not hemiclonic/unilateral); GTCS, generalized tonic–clonic seizures; H, hemiclonic; IS, infantile spasms; MA, myoclonic–astatic; MJ, myoclonic jerks; NCS, non-convulsive status epilepticus; SE, status epilepticus; SG, secondary generalization; T, tonic seizures; CGE, cryptogenic generalized epilepsy; CFE, cryptogenic focal epilepsy; LGS, Lennox–Gastaut syndrome; MAE, myoclonic–astatic epilepsy; SIMFE, severe infantile multifocal epilepsy; ID, intellectual disability; ND, not done.
The other four cases presented a mixed picture, but generalized spike wave and focal discharges were usually seen. The severity of the seizures varied with some only having generalized tonic–clonic seizures, which settled by adult life.
Cryptogenic focal epilepsy
Of 18 patients with cryptogenic focal epilepsy within this cohort of infantile epileptic encephalopathies, four (22%) had mutations (Table 1). Five cases presented with severe infantile multifocal epilepsy with developmental delay and are described later. Three had mutations: two (Patients 84 and 86) arose de novo (F575fsX622, R1596C) and one was maternally inherited. The latter (Patient 85) had a putative mutation (F1543S) that was highly conserved (Supplementary Fig. S1) and was carried by her unaffected mother, and may represent a susceptibility allele.
One (Patient 87) had recurrent febrile status epilepticus with onset at 18 months (Table 2). Twenty-four episodes of status epilepticus occurred, some with focal features with variable lateralization. MRI was normal. The patient died at 5 years due to complications of status epilepticus. He had a de novo missense SCN1A mutation (R1657H).
Severe infantile multifocal epilepsy
Five cases had this phenotype with seizure onset at a mean of 4 months. Of those with SCN1A mutations (Patients 84, 85 and 86, Table 2), onset occurred at mean of 5.5 months (4.5, 5 and 7 months) compared with 6- and 8-week onsets in the other two cases. Each child had multiple types of focal seizures with varying semiology. EEG studies showed abundant multifocal epileptiform activity typically with no (or exceptional) generalized or bilaterally synchronous discharges. The three patients with mutations had MRI brain studies; two showed mild atrophy. The remaining two had CT brain scans; one showed mild right sided atrophy.
Developmental delay became evident in all cases. In the two cases that were mutation negative, seizures began at 6 and 8 weeks concurrent with the recognition that developmental delay was present. In the three cases with SCN1A mutations, early development was normal with developmental slowing noted at the ages of 16 months, 3–4 years and 6 years even though seizure onset occurred at 4.5, 5 and 7 months, respectively (Table 2). Developmental outcome was poor with intellectual disability ranging from mild (one case, mutation positive: Patient 85), moderate (three cases, two had mutations: Patients 84 and 86) to severe (one case).
Other phenotypes
De novo SCN1A mutations were identified in 2/10 patients with MAE (Patients 89 and 90) and 1/12 patients with LGS (Patient 88) (Tables 1 and 2). No mutations were identified in patients with West syndrome, idiopathic spasms, early myoclonic encephalopathy, progressive myoclonic epilepsy, alternating hemiplegia of childhood or the 12 cases that could not be classified.
Discussion
The sodium channel alpha 1 subunit gene, SCN1A, is currently the most clinically relevant epilepsy gene. Mutations in SCN1A are an important cause of SMEI and SMEB and its subset ICEGTC (Claes et al., 2001; Mulley et al., 2005). Recently we showed that so-called ‘vaccine encephalopathy’ should be regarded as SMEI/SMEB on clinical and molecular grounds (Berkovic et al., 2006). Whilst SCN1A was originally associated with a small proportion of patients with the mild phenotypes characteristically seen in the GEFS+ syndrome (Escayg et al., 2000; Mulley et al., 2005), mutations within this gene have been identified far more often in patients with more severe forms of epilepsy. This study examines epileptic encephalopathies beginning early in life and expands the phenotypic spectrum of SCN1A defects beyond that previously recognized, to now include patients with cryptogenic generalized epilepsy and cryptogenic focal epilepsy.
The majority of mutations identified in the 90 children in this study were novel (72/90, 80%), whereas 18 (20%) had been previously published (Claes et al., 2001; Ohmori et al., 2002; Sugawara et al., 2002; Fujiwara et al., 2003; Nabbout et al., 2003a; Wallace et al., 2003; Fukuma et al., 2004; Mulley et al., 2005; Kearney et al., 2006; Mancardi et al., 2006; Marini et al., 2006). This expanded list of mutations, taken together with those reviewed by Mulley et al. (2005), provides an essential mutational database for use as an interpretative aid for diagnostic laboratories offering SCN1A mutation testing. Unlike some disorders where mutations are largely concentrated in ‘hot spots’, the mutations within SCN1A are widely distributed throughout the gene.
Parental DNA was available in 84% (76/90) of cases of which 96% (73/76) arose de novo and 4% (3/76) were familial. Familial SCN1A mutations have been previously reported in around 5% of SMEI where family members have mild GEFS+ phenotypes, as we observed here (Supplementary Fig. S2) (Fujiwara et al., 2003; Nabbout et al., 2003a; Mulley et al., 2005). In these probands, it is likely that their disorder has a multifactorial basis where SCN1A is a major but not the sole contributing gene. This would explain the marked disparity in phenotypic severity between the proband and their relatives. This model could explain probands 63, 82 and 85 where the parent was unaffected or had a mild phenotype. It is worth noting that these probands had a range of phenotypes including cryptogenic generalized and cryptogenic focal epilepsies.
Given the current state of knowledge, the majority of SCN1A mutations remain novel. This creates a challenge in determining whether new variants are pathogenic or not. Where the variant is de novo or results in truncation of the protein, then the likelihood of it being pathogenic is extremely high; 79 (88%) of our 90 positive cases fitted these criteria. In cases with missense changes, where DNA from parents is unavailable, or where an unaffected transmitting parent is identified, the case for pathogenicity rests on circumstantial evidence provided by evolutionary conservation of protein structure. Definitive functional studies are rarely available for this particular ion channel. In the four cases with two rare SCN1A variants, one of the two was assessed as more likely to be relevant to the observed phenotype. In cases where an unaffected transmitting parent is identified, these changes may be incidental benign variants, incompletely penetrant pathogenic variants or represent a susceptibility allele that contributes to the phenotype in a polygenic manner. Another alternative is that the variant has a major effect on the proband, whereas the transmitting parent has unrecognized protective molecular mechanisms.
SMEI
This study reinforces the high frequency of SCN1A mutations in patients with SMEI. The initial report described mutations in 7/7 cases (Claes et al., 2001). Subsequently, large series from a number of centres have reported mutations in 61–87% cases consistent with our finding of 79% reported here (Ohmori et al., 2002, 2003; Sugawara et al., 2002; Fujiwara et al., 2003; Fukuma et al., 2004). Lower mutation rates of 35% (33/93) and 33% (55/169) have been reported (Nabbout et al., 2003a; Suls et al., 2006) and of 33% (8/24) by our laboratory (Wallace et al., 2003). The latter study used single-strand conformation analysis for mutation detection, a rapid screening technology less sensitive than dHPLC used here. DHPLC has >96% sensitivity and specificity (Xiao and Oefner, 2001). Additional direct sequencing was performed in five cases (two negative). Fourteen of the remaining negative SMEI cases from our study were tested by dHPLC (3 cases) or direct sequencing (11 cases) here. Eight mutations were identified (two by dHPLC and six by sequencing), bringing the mutation rate to 16/24 (66%) for those cases reported in our original study (Wallace et al., 2003). Of our SCN1A mutation negative SMEI cases on dHPLC, 2 of 13 (15%) were subsequently found to have whole exon deletions detected by multiple ligase-dependent probe amplification (Mulley et al., 2006). Other SMEI cases lacking point mutations have been shown to have microdeletions including the SCN1A gene (Madia et al., 2006; Suls et al., 2006).
SMEB
We found 69% of our SMEB cases had SCN1A mutations. This figure is higher than the 26% reported by Fukuma et al. (2004) and more in keeping with the 88% mutation rate of Ohmori et al. (2003). The majority of SMEB mutations detected in this study were novel changes (17/25, 68%), with eight mutations being previously reported in patients with SMEI (Claes et al., 2001; Ohmori et al., 2002; Sugawara et al., 2002; Fujiwara et al., 2003; Nabbout et al., 2003a; Wallace et al., 2003; Fukuma et al., 2004; Kearney et al., 2006; Mancardi et al., 2006; Marini et al., 2006).
SMEB is distinguished from SMEI by the absence of specific features. The question of whether myoclonic seizures are an essential component of a SMEI phenotype remains controversial (Ogino et al., 1988; Commission on Classification and Terminology of the International League Against Epilepsy, 1989; Ohmori et al., 2003; Fukuma et al., 2004; Dravet et al., 2005). Dravet and colleagues observed that myoclonic seizures may be segmental or occur immediately prior to convulsive seizures and they postulate that subtle myoclonus may be missed (Dravet et al., 2005). Our data suggest that myoclonic seizures are not obligatory as three of four patients with an SMEI phenotype lacking only obvious myoclonic seizures (SMEB-M) carried a SCN1A mutation. Similarly, generalized spike-wave activity is considered the EEG hallmark of SMEI, but we found that 11/14 (79%) of our patients with a SMEI picture without demonstrated generalized spike-wave activity (SMEB-SW) had mutations.
Our findings in SMEB have important implications for the ‘lumpers and splitters’ debate. Whilst Ohmori and co-workers (2003) found a higher mutation rate in SMEB (88%) than SMEI (72%), our larger study shows the reverse. Moreover, three mutations are associated with both SMEI and SMEB (Patients 4 and 54, 8 and 59, 48 and 74) in this study. Similarly, eight cases have a mutation previously associated with the alternate phenotype (Supplementary Table). The recent ILAE classification proposal suggests the new name of Dravet syndrome for SMEI (Engel, 2001). In terms of clinical utility, we suggest that it may be more helpful to conceptualize SMEI and SMEB as a spectrum and incorporate both under the eponym of Dravet syndrome. This would also resolve the inaccuracy in terminology arising from the absence of myoclonic seizures in some cases of SMEI despite ‘myoclonic’ being part of the syndrome's name.
SCN1A mutations in SMEI and SMEB
Our data show similar results to those previously summarized in our review of SCN1A mutations (Mulley et al., 2005), with mutations comprising 43% (33/77) truncation and 43% (33/77) missense changes. The proportion of missense (39% versus 52%) and truncation (44% versus 40%) mutations is similar in SMEI and SMEB. Our new data fail to fully confirm previous observations of a predilection for missense mutations to occur in the ion channel pore region (Kanai et al., 2004), with only 15/33 (46%) in this region. Previous studies suggested clustering of missense mutations in SMEI in the S5–S5 loops of domain I and II (Mulley et al., 2005) but here, clustering in domain I was not seen (Fig. 1A).
No consistent pattern of clustering has emerged in SMEB although 18/25 mutations were located in the transmembrane domains (Fig. 1B). Here, clustering of mutations was noted in the S2–S4 transmembrane segments of domain I, in contrast to patterns seen previously where clustering in domain II was observed (Mulley et al., 2005). More data are required in order to establish if a true pattern of clustering exists.
Broader phenotypes of SCN1A mutations (Table 3)
The specific generalized epilepsy syndromes of MAE and LGS had a low yield of mutations with 2/10 and 1/12 positive cases respectively confirming that SCN1A is rarely associated with these syndromes (Wallace et al., 2001; Nabbout et al., 2003b; Ebach et al., 2005). The nosological boundaries between these disorders, SMEI, SMEB and other cryptogenic generalized epilepsies are blurred. Indeed, in the large group of patients with cryptogenic generalized epilepsy of early onset where a more specific syndromal diagnosis could not be reached, 6/25 had SCN1A mutations. Two patients had a phenotype with features similar to SMEI but had onset in early infancy with abnormal early development and a more severe course. Others had heterogeneous phenotypes of generalized epilepsy with intellectual disability including those previously recognized in GEFS+ families (Scheffer and Berkovic, 1997; Singh et al., 1999).
Epileptic encephalopathies with SCN1A mutations
| SMEI (n = 66) | SMEB (n = 36) | CGE (n = 25) | CFE (n = 13) | SIMFE (n = 5) | |
|---|---|---|---|---|---|
| Average age seizure onset (months) | 5.5 | 6 | 9.5 | 8 | 4 |
| Clinical features | |||||
| Hemiclonic and/or generalized convulsions | Always | Always | Often | Often | Often |
| Myoclonic seizures | Always | Often | Often | Occasional | Often |
| Other focal seizures | Often | Often | Occasional | Always | Always |
| Other generalized seizures | Often | Often | Often | Occasional | Rare |
| EEG | |||||
| Generalized spike wave | Always | Occasional | Often | Rare | No |
| Multifocal epileptiform activity | Occasional | Occasional | Occasional | Occasional | Always |
| SCN1A mutations | 52 (79%) | 25 (69%) | 6 (24%) | 1 (8%) | 3 (60%) |
| Truncation | 23 | 10 | – | – | 1 |
| Missense | 20 | 13 | 5 | 1 | 2 |
| Splice site | 9 | 2 | 1 | – | – |
| SMEI (n = 66) | SMEB (n = 36) | CGE (n = 25) | CFE (n = 13) | SIMFE (n = 5) | |
|---|---|---|---|---|---|
| Average age seizure onset (months) | 5.5 | 6 | 9.5 | 8 | 4 |
| Clinical features | |||||
| Hemiclonic and/or generalized convulsions | Always | Always | Often | Often | Often |
| Myoclonic seizures | Always | Often | Often | Occasional | Often |
| Other focal seizures | Often | Often | Occasional | Always | Always |
| Other generalized seizures | Often | Often | Often | Occasional | Rare |
| EEG | |||||
| Generalized spike wave | Always | Occasional | Often | Rare | No |
| Multifocal epileptiform activity | Occasional | Occasional | Occasional | Occasional | Always |
| SCN1A mutations | 52 (79%) | 25 (69%) | 6 (24%) | 1 (8%) | 3 (60%) |
| Truncation | 23 | 10 | – | – | 1 |
| Missense | 20 | 13 | 5 | 1 | 2 |
| Splice site | 9 | 2 | 1 | – | – |
SMEI, severe myoclonic epilepsy of infancy; SMEB, SMEI borderland; CGE, cryptogenic generalized epilepsy; CFE, cryptogenic focal epilepsy; SIMFE, severe infantile multifocal epilepsy.
Epileptic encephalopathies with SCN1A mutations
| SMEI (n = 66) | SMEB (n = 36) | CGE (n = 25) | CFE (n = 13) | SIMFE (n = 5) | |
|---|---|---|---|---|---|
| Average age seizure onset (months) | 5.5 | 6 | 9.5 | 8 | 4 |
| Clinical features | |||||
| Hemiclonic and/or generalized convulsions | Always | Always | Often | Often | Often |
| Myoclonic seizures | Always | Often | Often | Occasional | Often |
| Other focal seizures | Often | Often | Occasional | Always | Always |
| Other generalized seizures | Often | Often | Often | Occasional | Rare |
| EEG | |||||
| Generalized spike wave | Always | Occasional | Often | Rare | No |
| Multifocal epileptiform activity | Occasional | Occasional | Occasional | Occasional | Always |
| SCN1A mutations | 52 (79%) | 25 (69%) | 6 (24%) | 1 (8%) | 3 (60%) |
| Truncation | 23 | 10 | – | – | 1 |
| Missense | 20 | 13 | 5 | 1 | 2 |
| Splice site | 9 | 2 | 1 | – | – |
| SMEI (n = 66) | SMEB (n = 36) | CGE (n = 25) | CFE (n = 13) | SIMFE (n = 5) | |
|---|---|---|---|---|---|
| Average age seizure onset (months) | 5.5 | 6 | 9.5 | 8 | 4 |
| Clinical features | |||||
| Hemiclonic and/or generalized convulsions | Always | Always | Often | Often | Often |
| Myoclonic seizures | Always | Often | Often | Occasional | Often |
| Other focal seizures | Often | Often | Occasional | Always | Always |
| Other generalized seizures | Often | Often | Often | Occasional | Rare |
| EEG | |||||
| Generalized spike wave | Always | Occasional | Often | Rare | No |
| Multifocal epileptiform activity | Occasional | Occasional | Occasional | Occasional | Always |
| SCN1A mutations | 52 (79%) | 25 (69%) | 6 (24%) | 1 (8%) | 3 (60%) |
| Truncation | 23 | 10 | – | – | 1 |
| Missense | 20 | 13 | 5 | 1 | 2 |
| Splice site | 9 | 2 | 1 | – | – |
SMEI, severe myoclonic epilepsy of infancy; SMEB, SMEI borderland; CGE, cryptogenic generalized epilepsy; CFE, cryptogenic focal epilepsy; SIMFE, severe infantile multifocal epilepsy.
In patients classified as cryptogenic focal epilepsy, we identified a clinical subgroup who presented with a devastating multifocal epileptic encephalopathy. Of the five cases, three had SCN1A mutations. We designated this group severe infantile multifocal epilepsy (SIMFE) as onset is in the first year of life and multiple seizure types occur, with the most prominent being focal seizures. Multiple types of focal seizures occur including complex partial seizures of temporal lobe origin and hemiclonic seizures. Video-EEG telemetry showed that the variation in seizure semiology was not due to seizure spread patterns. Focal myoclonus may occur or even be brought out by specific anti-epileptic drugs known to exacerbate myoclonic seizures, such as vigabatrin. Patients may also have convulsive or non-convulsive status epilepticus, tonic seizures with focal features and tonic–clonic seizures. Interictal EEGs show abundant multifocal epileptiform discharges. These individuals do not have generalized spike-wave activity on EEG. Their MRI brain scans are normal or show non-specific features. They usually have normal early development followed by cognitive decline, with the refractory seizure disorder culminating in intellectual disability. Abnormal neurological signs such as ataxia and spasticity may evolve. The factor that distinguishes these children from SMEI is the absence of generalized absence and myoclonic seizures, generalized spike-wave activity on EEG, and that their cognitive decline may be later than the second year of life. These children had a severe, progressive and hitherto puzzling phenotype, where extensive investigations had been performed searching for an aetiology such as muscle biopsy, lumbar puncture and liver biopsy.
Similar cases are described in the literature by many authors (Noriega-Sanchez and Markand, 1976; Markand, 1977; Blume, 1978; Malik et al., 1989; Ohtsuka et al., 1990, 2000; Burnstine et al., 1991; Ohtahara et al., 1995; Nabbout and Dulac, 2003; Yamatogi and Ohtahara, 2003). Some clinicians regard this phenotype as being the later evolution of a ‘burnt out’ symptomatic generalized epilepsy, but these patients never have the EEG signature of generalized spike-wave activity. The phenotype could also be regarded as part of ‘severe epilepsy with multiple independent spike foci’ described by Ohtahara and colleagues where generalized minor seizures are also emphasized (Ohtsuka et al., 1990; Ohtahara et al., 1995; Yamatogi and Ohtahara, 2003, 2006). This group incorporates a heterogeneous array of causes such as tuberous sclerosis and birth asphyxia. In contrast, SIMFE encompasses those patients hitherto without a known cause, with 3/5 found to have mutations of SCN1A. SIMFE is an important group of patients with a devastating epileptic encephalopathy who are presently difficult to classify. The discovery of SCN1A mutations as the basis of their disorder avoids further potentially invasive investigations for alternative causes and assists in targeting therapy. For example, anti-epileptic drugs that exacerbate myoclonic seizures, such as vigabatrin and tiagabine, should be avoided.
This extensive study of the role of SCN1A mutations in epileptic encephalopathies beginning in the first year of life has, not surprisingly, expanded the phenotypic spectrum. Disorders are initially identified in a ‘pure cohort’ with a specific group of essential features. As the molecular basis is determined, phenotype–genotype correlation results in broadening of the phenotypic spectrum to include milder cases or seemingly unrelated disorders. This is just beginning to be possible in epileptology, as SCN1A is the first gene shown to have a role in epilepsies previously regarded as cryptogenic. An important finding is that children with an epileptic encephalopathy with multifocal features in the setting of normal MRI may have SCN1A mutations as may children with cryptogenic generalized epilepsy. A strong indicator for SCN1A analysis is an epileptic encephalopathy with seizure onset before 1 year of age, even if cognitive decline does not occur for several years thereafter. The social and economic benefit in making a definitive diagnosis in children with epileptic encephalopathies cannot be underestimated. Neurologists continue to perform investigations looking for an aetiology in children with cryptogenic encephalopathies such that establishing a definitive molecular diagnosis is cost-effective. More importantly, families are very grateful for a specific diagnosis especially with the treatment and genetic counselling implications that a SCN1A mutation carries.
Supplementary material
Supplementary material is available at Brain Online.
Acknowledgements
We thank the patients and their families for participating in this research. Funding was provided by the National Health and Medical Research Council of Australia, Thyne-Reid Charitable Trusts and Bionomics Ltd.
The Infantile Epileptic Encephalopathy Referral Consortium includes Kim Abbott, Ian Andrews, Barry Appleton, Andrew Bleasel, Neil Buchanan, Christopher Burke, Anne Bye, Carol Camfield, Peter Camfield, Gabriel Chow, Kevin Collins, Mark Cook, J. Helen Cross, Yanick Crow, M. Daniela D’Agostino, Martin Delatycki, Colin Dunkley, Joe Fawke, Colin Ferrie, Michael Geraghty, Gail Graham, Padraic Grattan-Smith, Elizabeth Hallam, Lorie Hamiwka, Anton Harding, Simon Harvey, Michael Hayman, Ian Hufton, Peter Humphries, Pierre Jacob, Raimond Jacquemard, David Jamison, Philip Jardine, Steve Jones, Daniel Keene, Kent Kelley, David Ketteridge, Andrew Kim, Sara Kivity, Chris Kneebone, Andrew Kornberg, Chris Lamb, Cecilie Lander, Tally Lerman-Sagie, Dorit Lev, Richard Leventer, Mark Mackay, Steven Malone, Jim Manson, Ailsa McLellan, Philip Moore, Lakshmi Nagarajan, Margot Nash, Marina Nikanorova, Doug Nordli, Mary O’Regan, Robert Ouvrier, Jayesh Patel, Clair Pridmore, Venkat Ramesh, David Reutens, Peter Rowe, Lloyd Shield, Paul Shillito, Lindsay Smith, Claire Spooner, Geoffrey Wallace, Nathan Watemberg, William Whitehouse and Elaine Wirrell.
References
Abbreviations:
- dHPLC
denaturing high performance liquid chromatography
- GEFS+
generalized epilepsy with febrile seizures plus
- ICEGTC
intractable childhood epilepsy with generalized tonic clonic seizures
- LGS
Lennox–Gastaut syndrome
- MAE
Myoclonic–astatic epilepsy
- PCR
polymerase chain reaction
- SCN1A
sodium channel alpha 1 subunit gene
- SMEB
SMEI-borderland
- SMEB-M
SMEI-borderland-myoclonic seizures
- SMEB-O
SMEI-borderland more than one feature
- SMEB-SW
SMEI-borderland-spike wave
- SMEI
severe myoclonic epilepsy of infancy


{kind=link}