




Abstract
Mild cognitive impairment (MCI), particularly the amnestic subtype (aMCI), is considered as a transitional stage between normal aging and a diagnosis of clinically probable Alzheimer's disease (AD). The aMCI construct is particularly useful as it provides an opportunity to assess a clinical stage which in most subjects represents prodromal AD. The aim of this study was to assess the progression of cerebral atrophy over multiple serial MRI during the period from aMCI to progression to AD. Thirty-three subjects were selected that fulfilled clinical criteria for aMCI and had three serial MRI scans: the first scan ∼3 years before the diagnosis of AD, the second scan ∼1 year before, and the third scan at the time of the diagnosis of AD. A group of 33 healthy controls were age and gender-matched to the study cohort. Voxel-based morphometry (VBM) was used to assess patterns of grey matter atrophy in the aMCI subjects at each time-point compared to the control group. Customized templates and prior probability maps were used to avoid normalization and segmentation bias. The pattern of grey matter loss in the aMCI subject scans that were 3 years before the diagnosis of AD was focused primarily on the medial temporal lobes, including the amygdala, anterior hippocampus and entorhinal cortex, with some additional involvement of the fusiform gyrus, compared to controls. The extent and magnitude of the cerebral atrophy further progressed by the time the subjects were 1 year before the diagnosis of AD. At this point atrophy in the temporal lobes spread to include the middle temporal gyrus, and extended into more posterior regions of the temporal lobe to include the entire extent of the hippocampus. The parietal lobe also started to become involved. By the time the subjects had progressed to a clinical diagnosis of AD the pattern of grey matter atrophy had become still more widespread with more severe involvement of the medial temporal lobes and the temporoparietal association cortices and, for the first time, substantial involvement of the frontal lobes. This pattern of progression fits well with the Braak and Braak neurofibrillary pathological staging scheme in AD. It suggests that the earliest changes occur in the anterior medial temporal lobe and fusiform gyrus, and that these changes occur at least 3 years before progression to the diagnosis of AD. These results also suggest that 3D patterns of grey matter atrophy may help to predict the time to the first diagnosis of AD in subjects with aMCI.
Introduction
Mild cognitive impairment (MCI) is generally considered a transitional stage between normal aging and a diagnosis of clinically probable Alzheimer's disease (AD). Amnestic MCI (aMCI) is a subtype in which subjects show early memory impairment, either with or without impairment of other cognitive domains, but do not fulfill criteria for dementia (Petersen et al., 1999; Petersen, 2004). Approximately 80% of subjects with aMCI go on to progress to a diagnosis of AD after a clinical follow-up of 6 years (Petersen, 2004). The MCI construct is therefore particularly useful as it provides an opportunity to assess a clinical stage which in most subjects represents prodromal AD.
MRI studies focus on patterns of cerebral atrophy in aMCI in order to identify the earliest changes in the brain associated with AD, and predict which subjects will progress to a diagnosis of AD. A number of region-of-interest (ROI) studies have found atrophy of the medial temporal lobe structures in MCI, including the hippocampus, entorhinal cortex, amygdala and parahippocampal gyrus, and found that atrophy of these structures can differentiate cross sectionally among controls, and subjects with MCI and AD (Xu et al., 2000; Du et al., 2001; Bottino et al., 2002; Pennanen et al., 2004; Wolf et al., 2004). The first studies demonstrating that quantitative MRI-based measures are associated with time to progression from MCI to AD focused on measures of the hippocampus (Jack et al., 1999; Visser et al., 1999). Subsequent studies demonstrated that quantification of atrophy of other medial temporal lobe structures, in addition to the hippocampus, is associated with a higher risk of subsequent progression to AD from MCI (Convit et al., 2000; Killiany et al., 2000; Dickerson et al., 2001; Killiany et al., 2002; Adak et al., 2004; DeCarli et al., 2004; deToledo-Morrell et al., 2004; Korf et al., 2004; Geroldi et al., 2006). Visual evaluation of CT (de Leon et al., 1993) or MRI (DeCarli et al., 2007) scans for medial temporal atrophy is also associated with greater risk of progression to AD in subjects with mild impairment. However these region-of-interest and visual evaluation techniques typically do not assess the full brain, and require a priori decisions concerning which structures to assess. Automated techniques have now been developed, such as voxel-based morphometry (VBM), which assess patterns of cerebral atrophy over the entire brain (Ashburner and Friston, 2000). Studies applying VBM to subjects with aMCI have identified similar patterns of medial temporal lobe atrophy but have also highlighted regions of atrophy in the inferior and lateral temporal lobes, the cingulate gyrus, and in the parietal and frontal lobes (Chetelat et al., 2002; Karas et al., 2004; Pennanen et al., 2005; Bell-McGinty et al., 2005). There is however a considerable variability across these studies in the degree of cortical involvement, most likely due to differences in disease stage, the clinical criteria used for the diagnosis of MCI, and the group size. In addition, there is variability in the time before the subjects with MCI progress to a diagnosis of AD. A couple of VBM studies have shown that subjects that remain stable for longer have less atrophy than those that progress more rapidly (Chetelat et al., 2005; Bozzali et al., 2006). These cross-sectional studies therefore allow us to infer which structures are involved earliest in AD, however they do not provide a direct assessment of change over time.
Longitudinal studies that assess serial scans provide more valuable information about how the patterns of cerebral atrophy change over time and will be important for future disease modifying trials. A number of studies have demonstrated elevated rates of cerebral atrophy in subjects with MCI compared to controls (Jack et al., 2000; Cardenas et al., 2003), and shown that the rates are higher in subjects which progress to AD (Jack et al., 2000, 2004; Chetelat et al., 2005; Jack et al., 2005; Stoub et al., 2005; Erten-Lyons et al., 2006). However, in order to assess disease progression patterns of atrophy need to be assessed over multiple different points in the disease course. One way to achieve this is to assemble independent groups of subjects that are at different stages of the disease, perhaps using a cognitive measure as a surrogate marker of disease stage (Mizuno et al., 2000; Wang et al., 2003). One study attempted to do this by assessing how the brain changes in groups of subjects with moderate AD, mild AD and in a group of subjects that were scanned before they became symptomatic (Scahill et al., 2002). They identified changes in the hippocampus in the presymptomatic subjects which spread to the inferolateral temporal lobes by the time subjects were mildly or moderately affected. However, the disadvantage of this approach is that normal inter-subject anatomic variability may be larger than the specific effect of disease stage on anatomy. This could be a particular problem in mildly affected subjects with MCI. The better approach would be to select a group of individuals that have multiple serial MRI. This would eliminate the confounding effect of inter-individual anatomic variability when assessing anatomic change attributable to disease progression. A number of ROI studies have used this approach to demonstrate increasing rates of atrophy over time in subjects that are at risk of developing familial AD (Fox et al., 1996; Schott et al., 2003; Ridha et al., 2006). However, no previous VBM studies have examined 3D patterns of cerebral atrophy over the entire brain with multiple (>2) time-points in subjects with aMCI.
The aim of this study was therefore to use VBM to assess the progression of cerebral atrophy over multiple serial MRI in a relatively large number of subjects with aMCI. Since we aimed to assess changes over time in prodromal AD we only selected subjects that had progressed to AD at follow-up, and then assessed the patterns of progression leading up to the point of the first diagnosis of AD. While we use the MCI nomenclature, others may view this as a progression along a continuum. This study aims to profile this progression using MRI.
Material and Methods
Subjects
Amnestic MCI subjects were identified from the Mayo Clinic Alzheimer's Disease Research Centre (ADRC) and Alzheimer's Disease Patient Registry (ADPR) in Rochester, Minnesota, USA. Informed consent was obtained for participation in the studies, which were approved by the Mayo Institutional Review Board. All subjects recruited into the ADRC and ADPR were followed prospectively and underwent approximately annual neurological, neuropsychological and neuroimaging assessments. These assessments included the Mini-Mental Status Examination (MMSE) (Folstein et al., 1975), the Clinical Dementia Rating sum of boxes (CDR-SOB) (Hughes et al., 1982) and the Dementia Rating Scale (DRS) (Mattis, 1976). Patients were diagnosed as having aMCI if they fulfilled the following criteria: (1) memory complaint, preferably corroborated by an informant; (2) memory impairment for age; (3) essentially normal general cognitive function; (4) generally preserved activities of daily living; (5) not demented (Petersen et al., 1999; Petersen, 2004). These well-established criteria have been used by our institution for many years and are essentially the same as those adopted by the National Institute on Aging (NIA) Alzheimer's Disease Centers Program and the Alzheimers Disease Neuroimaging Initiative (ADNI) (http://www.adni-info.org/images/stories/Documentation/adni_protocol_03.02.2005_ss.pdf). In all cases the diagnosis was made on clinical grounds without reference to MRI. Patients were reevaluated annually and the decision of whether subjects had progressed to clinically probable AD was made at a consensus committee meeting as previously described (Petersen et al., 2006). The diagnosis of probable AD was made according to NINCDS-ADRDA criteria (McKhann et al., 1984).
Apolipoprotein E testing was also performed. Blood was drawn from the study participants after receiving informed consent. DNA was amplified by means of polymerase chain reactions (Crook et al., 1994), and APOE genotyping was determined by technicians blinded to the clinical diagnosis.
Subjects were selected for this study if they fulfilled clinical criteria for aMCI (Petersen et al., 1999; Petersen, 2004) and had three serial MRI scans performed at 1.5 T. Each subject must have had two serial scans while carrying the diagnosis of aMCI and one scan at the point of the diagnosis of AD (i.e. a scan series of aMCI-aMCI-AD). We aimed to divide the aMCI scans into two groups based on the time that the scans were performed before conversion; a group of scans performed 1 year before the diagnosis of AD and a group of scans performed 3 years before the diagnosis of AD. To account for the inevitable variability in scan intervals across subjects we had to set specific time interval windows to define each of these groups. For example, although the study design plan may have been to scan each subject at precisely yearly intervals, the actual scan dates may have been a few months or more in some cases from this target due to difficulties in patient scheduling. In addition, in order to provide enough separation between the three groups’ the intervals between serial scans from each individual had to be greater than 9 months. Therefore, in order to capture scans that were binned into the ‘1 year before the diagnosis of AD’ time window, the minimum time before the diagnosis of AD was set to 9 months. The maximum time before the diagnosis of AD was set to 18 months in order to capture scan intervals that were slightly greater than 1 year. There was more variability in the time that the scan furthest from the diagnosis of AD was performed, therefore the scan window was larger than the window set for the 1 year scans. The scans selected for the ‘3-year before the diagnosis of AD group’ had to have a minimal interval before the diagnosis of AD of greater than 18 months. Then in order to centre the variability around 3 years the maximum interval was set at 54 months. Therefore we have a window of 18 months either side of 3 years. The clinical history and MRI scans were reviewed in all cases. Subjects with structural abnormalities that could produce cognitive impairment, or who had treatments or concurrent illnesses interfering with cognitive function either at baseline or during follow-up were not included in this study. MRI scans were rejected if they were corrupted by scan artefacts, such as movement or susceptibility artefacts, which could prevent accurate segmentation.
Sixty-one subjects were identified from the ADRC and ADPR that fulfilled clinical criteria for amnestic aMCI and had three serial MRI scans at the correct clinical diagnoses, i.e. two serial MRI while the subject was diagnosed as aMCI, and one at the time of the diagnosis of AD (i.e. aMCI-aMCI-AD). Of these, 11 subjects were excluded due to medical exclusions, nine were excluded due to unusable MRI and eight were excluded because the MRI scans did not fit into our specific time windows. Therefore, 33 subjects with three serial scans were identified that fulfilled our inclusion criteria. The median time interval between the first MRI and the diagnosis of AD was 3 years (range 1.7–4.2 years), and the median interval between the second MRI and the diagnosis of AD was 1 year (range 0.8–1.5 years). The scan intervals for each of the 33 subjects with aMCI are shown in Fig. 1.
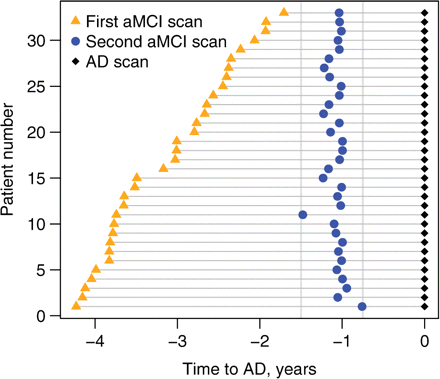
Time-line representing the time of each MRI scan for each individual (n = 33) relative to progression from aMCI to a clinical diagnosis of AD. The black diamonds represent the scan at the time of the diagnosis of AD. The blue circles represent the scans classified as being 1 year (9–18 months) before the diagnosis of AD. The orange triangles represent the scans classified as being 3 years (18–51 months) before the diagnosis of AD.
Each subject that was included in the analysis was age and gender-matched to a control subject. The matching was performed to the middle scan for each aMCI subject. The date/year that the scans were performed were also matched in an attempt to control for any temporal fluctuations associated with different scanner platform versions. All the control subjects were prospectively recruited via the same mechanism as the aMCI subjects into the Mayo Clinic ADRC, or the ADPR, and were identified from the ADRC/ADPR database. Control subjects were cognitively normal individuals that had been seen in internal medicine for routine physical examinations and asked to enroll in the ADRC and ADPR. All subjects were then evaluated by a neurologist to verify the normal diagnosis. Controls were identified as individuals who (a) were independently functioning community dwellers, (b) did not have active neurological or psychiatric conditions, (c) had no cognitive complaints, (d) had a normal neurological and neurocognitive examination and (e) were not taking any psychoactive medications in doses that would affect cognition.
Image acquisition
T1-weighted 3D volumetric spoiled gradient echo (SPGR) sequences with 124 contiguous partitions and 1.6 mm slice thickness (22 × 16.5 or 24 × 18.5 cm2 FOV, 25° flip angle) were performed and used for analysis. An identical scan acquisition protocol was used for all scans. Different scanners were used, but all were GE Signa 1.5 T with body resonance module gradient sets and transmit-receive single channel head coils. All scanners undergo a standardized quality control calibration procedure daily, which monitors geometric fidelity over a 200 mm volume along all three cardinal axes, signal-to-noise and transmit gain, and maintains the scanner within a tight calibration range.
Image analysis
An optimized method of VBM, implemented using SPM2 (http://www.fil.ion.ucl.ac.uk/spm), was used to assess patterns of grey matter atrophy in each of the different aMCI scan groups (Ashburner and Friston, 2000; Senjem et al., 2005). There were three comparisons of interest: (1) the group of scans ∼3 years before the diagnosis of AD versus the control group, (2) the group of scans ∼1 year before the diagnosis of AD versus the control group and (3) the group of scans at the time of the diagnosis of AD versus the control group. Note that a single set of scans from the same control group is used in all three comparisons. The VBM processing steps and analysis were therefore performed separately for the images for each of these comparisons as if they were separate studies.
Customized templates and prior probability maps (priors) were created in order to reduce any potential normalization bias across the disease groups. Separate customized templates and priors were created for each of these three comparisons. Therefore in each comparison, all subjects were registered to the Montreal Neurological Institute (MNI) template using a 12 degrees of freedom (dof) affine transformation and segmented into grey matter (GM), white matter (WM) and CSF using MNI priors. GM images were normalized to the MNI GM prior using a non-linear discrete cosine transformation (DCT). The normalization parameters were applied to the original whole head and the images were segmented using the MNI priors. Average images were created of whole head, GM, WM and CSF, and smoothed using 8 mm full-width at half-maximum (FWHM) smoothing kernel.
The VBM processing was similarly performed separately for each of the three comparison groups. All images required for the comparison were registered to the comparison-specific customized whole brain template using a 12 dof affine transformation and segmented using the comparison-specific customized priors. The GM images were normalized to the comparison-specific custom GM prior using a non-linear DCT. The normalization parameters were then applied to the original whole head and the images were segmented once again using the comparison-specific customized priors. All images were modulated and smoothed with an 8 mm FWHM smoothing kernel. In addition, a re-initialization routine was implemented. This uses the parameters from the initial normalization to the MNI template (performed to generate the customized template) to initialize the normalization to the custom template (Senjem et al., 2005). Grey matter differences were assessed using the general linear model on a voxel basis at a statistical threshold of P < 0.01 after correction for multiple comparisons using the false discovery rate (FDR).
Statistics
Two-sided two-sample Wilcoxon rank-sum tests were used to compare the aMCI group to the matched cognitively healthy subjects on age and years of education. A chi-square test was used to compare the gender ratio and the proportion of apolioprotein epsilon 4 (APOE ε 4) carriers between the groups. To estimate the rate of annual decline in cognition among the aMCI group, we calculated a least squares slope for each patient for MMSE, CDR and DRS. We estimated the average annual cognitive decline by taking the median of these slope estimates. We report medians and use non-parametric methods due to skewness in the numeric clinical variables.
Results
Subject demographics
The demographics for the control and aMCI subjects are shown in Table 1. By design, there was no statistical difference in age or gender ratio across the groups. There was also no statistical difference in years of education. The frequency of APOE ε4 carriers was however significantly higher in the aMCI group compared to the control group; with a frequency of 71.9% in the aMCI subjects compared to only 18.2% in the control population. Table 2 provides cognitive test data for the aMCI subjects at each time-point. There was a decrease in MMSE and DRS, and increase in CDR-SOB, over time in the aMCI subjects. The MMSE decreased at a rate of 0.9 points per year, the CDR-SOB increased at a rate of 0.7 points per year, and the DRS decreased at a rate of 3 points per year over the study period. Six of these subjects have since come to autopsy. The median time between the last MRI and death was 3 years (range 1–7). All six subjects had AD-type pathology of varying severity. Secondary pathologies included the presence of limbic Lewy Bodies in one case, and agyrophilic grains in another.
Subject demographics for the aMCI progressors and controls
| aMCI progressors (n = 33) | Controls (n = 33) | P values | |
|---|---|---|---|
| No. of females | 19 (58) | 19 (58) | – |
| Median (range) age, yearsa | 78 (65, 92) | 78 (63, 93) | 0.97b |
| Median (range) education, years | 16 (7, 20) | 13 (8, 18) | 0.23b |
| No. of APOE ε4 carriers (%) | 23 (71.9) | 6 (18.2) | <0.001c |
| aMCI progressors (n = 33) | Controls (n = 33) | P values | |
|---|---|---|---|
| No. of females | 19 (58) | 19 (58) | – |
| Median (range) age, yearsa | 78 (65, 92) | 78 (63, 93) | 0.97b |
| Median (range) education, years | 16 (7, 20) | 13 (8, 18) | 0.23b |
| No. of APOE ε4 carriers (%) | 23 (71.9) | 6 (18.2) | <0.001c |
aAge at time of the scan 9–18 months prior to the diagnosis of AD.
bKruskal Wallis test was performed across groups.
cChi-square test was performed across groups.
Subject demographics for the aMCI progressors and controls
| aMCI progressors (n = 33) | Controls (n = 33) | P values | |
|---|---|---|---|
| No. of females | 19 (58) | 19 (58) | – |
| Median (range) age, yearsa | 78 (65, 92) | 78 (63, 93) | 0.97b |
| Median (range) education, years | 16 (7, 20) | 13 (8, 18) | 0.23b |
| No. of APOE ε4 carriers (%) | 23 (71.9) | 6 (18.2) | <0.001c |
| aMCI progressors (n = 33) | Controls (n = 33) | P values | |
|---|---|---|---|
| No. of females | 19 (58) | 19 (58) | – |
| Median (range) age, yearsa | 78 (65, 92) | 78 (63, 93) | 0.97b |
| Median (range) education, years | 16 (7, 20) | 13 (8, 18) | 0.23b |
| No. of APOE ε4 carriers (%) | 23 (71.9) | 6 (18.2) | <0.001c |
aAge at time of the scan 9–18 months prior to the diagnosis of AD.
bKruskal Wallis test was performed across groups.
cChi-square test was performed across groups.
Cognitive test data for the aMCI progressors subjects at each serial MRI time-point
| aMCI (3 years before the diagnosis of AD) | aMCI (1 year before the diagnosis of AD) | AD (time of the diagnosis of AD) | Slope trend median (95% CI) | |
|---|---|---|---|---|
| Median (range) MMSE score | 27 (24, 30) | 25 (22, 29) | 24 (14, 30) | −0.9 (−1.4, −0.7) |
| Median (range) CDR-SOB | 1.0 (0.0, 6.0) | 2.0 (0.5, 4.5) | 3.5 (1.5, 12) | 0.7 (0.6, 1.1) |
| Median (range) DRS score | 132 (116, 141) | 125 (106, 136) | 120 (99, 135) | −3.0 (−4.3, −2.1) |
| aMCI (3 years before the diagnosis of AD) | aMCI (1 year before the diagnosis of AD) | AD (time of the diagnosis of AD) | Slope trend median (95% CI) | |
|---|---|---|---|---|
| Median (range) MMSE score | 27 (24, 30) | 25 (22, 29) | 24 (14, 30) | −0.9 (−1.4, −0.7) |
| Median (range) CDR-SOB | 1.0 (0.0, 6.0) | 2.0 (0.5, 4.5) | 3.5 (1.5, 12) | 0.7 (0.6, 1.1) |
| Median (range) DRS score | 132 (116, 141) | 125 (106, 136) | 120 (99, 135) | −3.0 (−4.3, −2.1) |
Note: MMSE = Mini-Mental Status Examination; CDR-SOB = Clinical Dementia Rating sum of boxes; DRS = Dementia Rating Scale; CI = confidence interval.
Cognitive test data for the aMCI progressors subjects at each serial MRI time-point
| aMCI (3 years before the diagnosis of AD) | aMCI (1 year before the diagnosis of AD) | AD (time of the diagnosis of AD) | Slope trend median (95% CI) | |
|---|---|---|---|---|
| Median (range) MMSE score | 27 (24, 30) | 25 (22, 29) | 24 (14, 30) | −0.9 (−1.4, −0.7) |
| Median (range) CDR-SOB | 1.0 (0.0, 6.0) | 2.0 (0.5, 4.5) | 3.5 (1.5, 12) | 0.7 (0.6, 1.1) |
| Median (range) DRS score | 132 (116, 141) | 125 (106, 136) | 120 (99, 135) | −3.0 (−4.3, −2.1) |
| aMCI (3 years before the diagnosis of AD) | aMCI (1 year before the diagnosis of AD) | AD (time of the diagnosis of AD) | Slope trend median (95% CI) | |
|---|---|---|---|---|
| Median (range) MMSE score | 27 (24, 30) | 25 (22, 29) | 24 (14, 30) | −0.9 (−1.4, −0.7) |
| Median (range) CDR-SOB | 1.0 (0.0, 6.0) | 2.0 (0.5, 4.5) | 3.5 (1.5, 12) | 0.7 (0.6, 1.1) |
| Median (range) DRS score | 132 (116, 141) | 125 (106, 136) | 120 (99, 135) | −3.0 (−4.3, −2.1) |
Note: MMSE = Mini-Mental Status Examination; CDR-SOB = Clinical Dementia Rating sum of boxes; DRS = Dementia Rating Scale; CI = confidence interval.
Image analysis
Three years before the diagnosis of AD
The pattern of grey matter loss in the MCI subject scans that were approximately 3 years before the diagnosis of AD was focused primarily on the medial temporal lobes, including the left amygdala, and bilateral anterior hippocampus, entorhinal cortex and fusiform gyrus (corrected for multiple comparisons, P < 0.01) (Fig. 2). The pattern of loss was bilateral although slightly greater on the left. It is notable that the posterior hippocampus was relatively spared and no significant grey matter loss was observed outside of the temporal lobes.
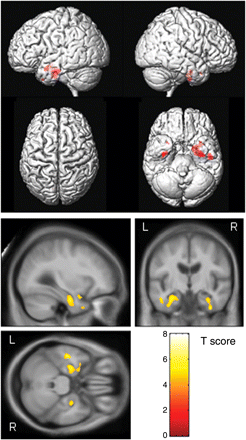
Patterns of grey matter atrophy in the aMCI progressors ∼3 years (18–54 months) before progression to AD. The results are shown on a 3D surface render (top) and overlaid on representative axial, coronal and sagittal slices (bottom). L = left; R = right.
One year before the diagnosis of AD
The MCI scans that were ∼1 year before the diagnosis of AD showed greater grey matter loss compared to controls than observed in the scans performed 3 years before the diagnosis of AD. The brunt of the loss was still focused on the medial and inferior temporal lobes, although the extent and magnitude of the temporal lobe loss was greater (corrected P < 0.01, Fig. 3). The amygdala, hippocampus, inferior temporal gyrus, entorhinal cortex, fusiform gyrus and the anterior temporal lobe were all involved, but there was also some involvement of the anterior middle temporal gyrus. The grey matter loss in the temporal lobes also extended into the posterior temporal lobe; including the whole extent of the hippocampus. As before, the pattern of loss was bilateral although slightly greater on the left. Regions of loss were also observed bilaterally in the parietal lobe yet the frontal lobes were relatively spared.
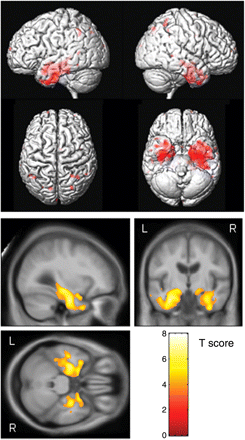
Patterns of grey matter atrophy in the aMCI progressors ∼1 year (9–18 months) before progression to AD. The results are shown on a 3D surface render (top) and overlaid on representative axial, coronal and sagittal slices (bottom). L = left; R = right.
At the diagnosis of AD
The patterns of grey matter loss identified at the time of progression from aMCI to a diagnosis of AD were strikingly more widespread than those observed 1 year prior to conversion. Severe grey matter loss was observed throughout the temporal lobes, in the temporoparietal association neocortex, and in the frontal lobes (corrected P < 0.01, Fig. 4). All temporal lobe gyri were involved, although there was a relative sparing of the posterior superior temporal gyrus. The left temporal lobe was still involved to a greater extent than the right. Loss in the parietal lobe was relatively symmetric. The grey matter loss observed in the frontal lobes was predominantly located in the anterior frontal lobe and the superior frontal gyri. Grey matter loss was also observed in the midbrain.
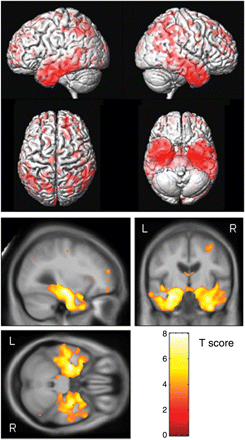
Patterns of grey matter atrophy in the aMCI progressors at the time of a diagnosis of AD. The results are shown on a 3D surface render (top) and overlaid on representative axial, coronal and sagittal slices (bottom). L = left; R = right.
Discussion
In this study we illuminate the 3D progression of grey matter atrophy over multiple MRI scans in individuals with aMCI that progress to a diagnosis of AD. The results showed progression in both the severity and distribution of atrophy over time, and demonstrated that the earliest changes that can be detected by MRI occur in anterior temporal regions.
Grey matter losses were first identified in the medial and inferior temporal lobes, including the amygdala, hippocampus, entorhinal cortex and fusiform gyrus, ∼3 years before progression to AD. Interestingly the grey matter loss was predominantly located in the anterior regions of the temporal lobe, with relative sparing of the posterior hippocampus. These structures are known to be some of the first to be involved in the progression of AD pathology (Braak and Braak, 1996) and have been implicated in previous VBM studies of subjects with MCI (Chetelat et al., 2002; Karas et al., 2004; Chetelat et al., 2005; Pennanen et al., 2005; Bozzali et al., 2006; Whitwell et al., in press). Region-of-interest studies have also shown that medial temporal or a combination of medial and inferior temporal lobe measurements can provide the best discrimination between subjects with AD and controls, and hence concluded that these are likely to be the first regions of loss in MCI (Jack et al., 1992, 1997; Convit et al., 2000; Morys et al., 2002). This is the first study to investigate the time-dependent evolution of 3D atrophy patterns leading up to a diagnosis of AD through serial MRI. It is notable that at 3 years prior to a diagnosis of AD the atrophy did not spread outside the temporal lobes, with no involvement of the parietal or frontal regions. The extent and magnitude of the cerebral atrophy further progressed by the time the subjects were 1 year before the diagnosis of AD. Atrophy in the temporal lobes spread to include the middle temporal gyrus, and extended into more posterior regions of the temporal lobe to include the entire extent of the hippocampus. The parietal lobe also started to become involved, although the frontal lobe was still relatively spared. A number of previous studies have similarly failed to find atrophy of the frontal lobes in subjects with aMCI (Chetelat et al., 2002; Hirata et al., 2005; Pennanen et al., 2005).
By the time the subjects had progressed to a clinical diagnosis of AD the pattern of cerebral atrophy detected on MRI had become dramatically more widespread with more severe involvement of the medial temporal lobes and the temporoparietal association cortices and, for the first time, substantial involvement of the frontal lobes. These regions are all typically involved in AD (Fox et al., 1996; Jack et al., 1997; Baron et al., 2001; Frisoni et al., 2002; Matsuda et al., 2002). These widespread patterns of loss likely correspond to the worsening cognitive functioning that led to the progression to AD. A couple of cross-sectional VBM studies have compared subjects with AD to those with aMCI and have similarly shown greater atrophy in the temporal, parietal and frontal lobes in AD than aMCI (Chetelat et al., 2002; Karas et al., 2004). In addition, a recent study by Chetelat and colleagues (2005) investigated the longitudinal patterns of atrophy associated with progression from MCI to AD over an 18-month period in seven subjects with MCI (Chetelat et al., 2005). Similar to our study they noted an increase in atrophy detected on MRI in the medial and inferior temporal lobes with progression, however they also identified increases in the posterior cingulate and precuneus. Differences across the studies are most likely due to the different methodology. The Chetelat study evaluated two scans in each subject and performed a non-linear registration between the baseline and follow-up scans and calculated changes directly between the two time-points. In contrast, in our study, which had three serial MRI, the patterns of atrophy at each time-point were determined by comparison to a common control group.
The pattern of progression identified in this study fits nicely with the proposed pathological staging scheme in AD (Braak and Braak, 1996). Neurofibrillary tangles first occur in the entorhinal cortex and the hippocampus (transentorhinal stages I–II), before spreading out into the amygdala and basolateral temporal lobe (limbic stages III–IV) and then into the isocortical association areas (isocortical stages V–VI). A pathological diagnosis of high-probability AD is given at Braak 5 and 6 when isocortical areas are involved. This fits with our patterns that show that although there is a little parietal involvement at MCI, a dramatic increase in cortical involvement occurs at the time of progression to AD. The patterns of atrophy observed at each disease stage were also bilateral, although showed greater involvement of the left hemisphere. The left-sided predominance may reflect the fact that the memory tests used for diagnosis were verbally weighted. Many previous studies have similarly shown left-sided patterns of cerebral atrophy in AD (Baron et al., 2001; Boxer et al., 2003; Karas et al., 2003).
The hippocampus showed progressive atrophy throughout the disease course, with the severity of hippocampal loss detected on MRI increasing at each time-point. These results concord with previous studies that have shown progressive hippocampal atrophy over time and with disease severity in subjects with AD (Fox et al., 1996; Jack et al., 1997). Rates of hippocampal atrophy have also been shown to increase as the disease progresses from control to MCI to AD (Jack et al., 2000, 2004; Ridha et al., 2006). VBM studies have similarly shown a greater degree of hippocampal atrophy in subjects with AD compared to MCI (Karas et al., 2004), which argues against the suggestion that hippocampal atrophy reaches a plateau (Chetelat et al., 2002). Interestingly the grey matter loss detected on MRI was predominantly located in the anterior regions of the hippocampus 3 years before the diagnosis of AD, and then progressed to involve the posterior hippocampus by 1 year before the diagnosis of AD. In concordance with these results previous studies have demonstrated that the hippocampal head shows greater atrophy than the body or tail in subjects with AD (Chang et al., 1992; Jack et al., 1997). In addition, two studies have demonstrated similar trends by assessing AD subjects with varying CDR scores. Mizuno and colleagues (2000) showed that the anterior portions of hippocampus atrophied in subjects at a mild CDR of 0.5, while atrophy of the posterior portions of the hippocampus was only found in subjects with a more moderate CDR of 2–3 (Mizuno et al., 2000). Similarly, Wang and colleagues (2003) demonstrated inward deformation of the hippocampal head in subjects with a CDR of 0.5, while the lateral regions of the hippocampus became involved ∼2 years later (Wang et al., 2003). These results all suggest that the anterior portion of the hippocampus is more susceptible to degenerative change than the posterior portion. This is however the first study to demonstrate this progression in MCI using multiple serial MRI scans from the same individuals.
Contrary to other studies on AD we failed to find any involvement of the posterior cingulate at either the MCI or AD time-points. Posterior cingulate dysfunction is a common finding in functional imaging studies of AD (Minoshima et al., 1997). MRI studies have however shown mixed results concerning involvement of the posterior cingulate, with some showing atrophy in AD (Baron et al., 2001; Frisoni et al., 2002; Matsuda et al., 2002; Boxer et al., 2003; Karas et al., 2003) and MCI subjects (Chetelat et al., 2002; Scahill et al., 2002; Karas et al., 2003; Shiino et al., 2006), while others have failed to find any involvement in mild AD (Busatto et al., 2003) and MCI (Pennanen et al., 2005). The degree of atrophy of the posterior cingulate and precuneus has been shown to vary dependent on age at onset (Vogt et al., 1998; Frisoni et al., 2005; Ishii et al., 2005; Shiino et al., 2006), with little or no involvement in subjects with an age of onset over 65 years. Posterior cingulate atrophy has also been observed to occur in normal aging (Shiino et al., 2006), therefore the age-matching between controls and disease subjects could be crucial. Our subjects were over 65 years of age and were all well matched to controls. However, since the MRI scans in this study were performed in aMCI and in very mild AD it is possible that the posterior cingulate will become involved later in the disease course. We also failed to find any involvement of subcortical structures. Some grey matter loss was observed surrounding the lateral ventricles at the time of the diagnosis of AD but this is most likely due to mis-segmentation of the periventricular white matter as a result of ventricular expansion. Regions surrounding the temporal horn of the lateral ventricle, such as the hippocampus and amygdala, could also be affected by this mis-segmentation, although it is unlikely to explain the severe degree of loss identified in these areas.
The strength of this study is that we have a relatively large number of clinically well-characterized subjects where each have three serial MRI that span well-defined disease stages. This allows us to investigate the progression of atrophy from 3 years before the diagnosis of AD to the time of the diagnosis of AD. Because the same group of subjects was followed longitudinally we were able to visualize the anatomic change attributable to disease progression free of the confounding effects of inter-individual anatomic variability—i.e. each person's initial scan served as his/her own baseline. Likewise, by generating the three group-wise comparisons with the same set of control scans in each contrast, we were able to eliminate anatomic variability in the reference group that might have obscured findings had we used three different sets of control scans. A possible limitation of this design is that age-related changes could have influenced the patterns of atrophy observed at the first and third time-points, since the control subjects were matched by age to the second time-point. However the difference in expected atrophy in controls over only a few years is likely to be minimal. In addition, the fact that we required three good quality MRI scans at specific time intervals means we would by definition be excluding subjects who were unable to cooperate for serial MRI due to rapidly progressive dementia. This may have led to an underestimation in the degree of cerebral atrophy at each stage, since the largest volume losses should occur in patients who were the rapid clinical decliners. Also, since there is an inevitable variability in scan interval across subjects, and due to unusable MRI, the group of scans 3 years before the diagnosis of AD had a larger variation in time before progression (ranging from 4.2 to 1.7 years) than the 1 year before the diagnosis of AD group. However, a good time separation was maintained between the groups. The VBM technique is also limited by the fact that multiple statistical comparisons are performed across the brain which may increase the chance of false positive results. We and others have corrected for this problem by applying the widely used false discovery rate (Genovese et al., 2002; McMillan et al., 2004; Teipel et al., 2004) correction for multiple comparisons.
The study was limited by the lack of pathological confirmation in all subjects. However, all six subjects that had come to autopsy showed AD-type pathology and a recent study from our institution showed that a high proportion of aMCI subjects that progress clinically to AD will have AD on pathology (Jicha et al., 2006). In addition, the frequency of APOE e4 carriers was high in the aMCI subjects providing further evidence that these subjects are likely to have AD at autopsy.
These results therefore demonstrate that VBM can be used to map the 3D progression of grey matter loss in groups of subjects with aMCI. If an image analysis method that provided single subject classification were employed (Burges, 1998; Davatzikos et al., 2006) these patterns of loss may help to predict time from progression in subjects with aMCI.
Acknowledgements
This study was supported by grants P50 AG16574, U01 AG06786 and R01 AG11378 from the National Institute on Aging, Bethesda MD, the generous support of the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, USA. D.S.K. has been a consultant to GE HealthCare, GlaxoSmithKline and Myriad Pharmaceuticals, has served on a Data Safety Monitoring Board for Neurochem Pharmaceuticals, and is an investigator in a clinical trial sponsored by Elan Pharmaceuticals. R.C.P. has been a consultant to GE Healthcare and is on a Treatment Effects Monitoring Committee for a clinical trial sponsored by Elan Pharmaceuticals. B.B. is an investigator in a clinical trial sponsored by Myriad Pharmaceuticals. We would also like to acknowledge Dr Dennis Dickson, Dr Joseph Parisi and Dr Keith Josephs for conducting and reviewing the pathological analyses.
References
Abbreviations:
- CDR
Clinical Dementia Rating
- DRS
Dementia Rating Scale
- MCI
mild cognitive impairment
- MMSE
Mini-Mental Status Examination


{kind=link}
{kind=link}
{kind=link}
{kind=link}