




Abstract
Argyrophilic grain disease (AGD) is a common sporadic neurodegenerative disease of old age characterized by the presence of argyrophilic grains (AGs)—dendritic-derived appendages as revealed with the Golgi method—together with pre-tangle neurons in the limbic system, which accounts for about 5% of all demented cases. AGs and pre-tangle neurons contain hyperphosphorylated 4R tau. This is associated with a typical 64 kDa and 68 kDa pattern, but also accompanied by tau truncated forms of low molecular mass, probably resulting from thrombin-mediated proteolysis. Hyperphosphorylated tau also accumulates in oligodendroglial-coiled bodies and in limbic astrocytes. Ballooning neurons in the amygdala are non-specific accompanying abnormalities. A new proposal for AG distribution considers four stages. Clinical symptoms largely depend on the extension of AGs together with the very common associated tauopathies, mainly Alzheimer's disease, progressive supranuclear palsy, corticobasal degeneration and synucleinopathies. Pathogenesis of AG and related lesions herein proposed includes oxidative stress that is followed by increased expression of oxidative response markers, and activation of stress kinases (stress activated protein kinase and p38). These kinases together with glycogen synthase kinase 3β co-localize with hyperphosphorylated tau deposits in neurons and glial cells, thus indicating a link between oxidative stress and tau phosphorylation in AGD. Hyperphosphorylated tau, in turn, co-localizes with p62/sequestosome 1 and ubiquitin, thus pointing to activation of protein aggregation and protein degradation pathways, respectively. Finally, AGs and tangles co-localize with mutant ubiquitin (UBB+1) resulting from molecular misreading of mRNA, thus supporting proteasome function impairment and, therefore, impelling accumulation of hyperphosphorylated tau in AGs and tangles. The sequestration of active kinases in AGs and tangles is an additional local cause of tau hyperphosphorylation.
Introduction
Argyrophilic grain disease (AGD) was first described as a degenerative disease characterized by argyrophilic grains (AGs) in the entorhinal cortex, hippocampus, amygdala and neighbouring temporal cortex in a subset of patients who had suffered from adult onset dementia (Braak and Braak, 1987, 1989).
Although the seminal descriptions emphasized the lack of Alzheimer changes, subsequent studies have shown frequent association of AGD with other degenerative diseases of the nervous system, including Alzheimer's disease (AD), Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), dementia with only tangles, Creutzfeldt–Jakob disease (CJD) and α-synucleinopathies such as Lewy body diseases [Parkinson's disease(PD) and Dementia with Lewy bodies (DLB)] and multiple system atrophy (Masliah et al., 1991; Ikeda et al., 1995; de Vos et al., 1996; Martinez-Lage and Muñoz, 1997; Braak and Braak, 1998; Jellinger, 1998; Jellinger and Bancher, 1998; Kiwashima et al., 1999; Seno et al., 2000; Togo et al., 2002; Ferrer et al., 2003). Rare cases have been reported with combined association of AGD, Lewy body disease and motor neuron disease (Liang et al., 2005; Klos et al., 2005). As in AD, Lewy bodies are frequent in the amygdala in AGD (Popescu et al., 2004). Finally, hippocampal sclerosis may be found in combination with AGD, as it occurs in other cases of dementia with tauopathy (Beach et al., 2003; Probst et al., 2007).
Together, these observations have raised cautionary comments about the reality of AGD as a distinct entity (Martinez-Lage and Munoz, 1997; Ikeda et al., 2000), while the opposing view is defended by others (Tolnay et al., 2003; Tolnay and Clavaguera, 2004).
AGs are not uncommon structures in aged human brains (Davis et al., 1997). Their presence has been estimated at 5–9% in adult autopsy series (Braak and Braak, 1998; Tolnay and Clavaguera, 2004). In our series of 1000 consecutive autopsy cases from an adult general hospital, the percentage of cases with AGs was 4%. There is a general agreement that the incidence of AGD increases with age (Braak and Braak, 1998; Tolnay and Clavaguera, 2004). This may explain AGs in 43% of cases in a series of very aged patients (Saito et al., 2002) and the occurrence of AGD in 10 of 32 centenarians (Ding et al., 2006). The mean age of onset is about 75–80 years (Braak and Braak, 1998; Jellinger 1998; Tolnay et al., 2001; Tolnay et al., 2003). In our series, the distribution of AGD for ages was: younger than 60 years, 10%; between 61 and 70, 17%; between 71 and 80, 30%; and older than 80 years, 43%. Both genders are equally affected.
The cause of AGD is not known. The disease appears to be sporadic. Genetic studies have failed to discover a sustained link of AGD with a particular gene locus. Interestingly, a single case bearing the MAPT S305I mutation had AGD-like neuropathology (Kovacs et al., 2007). The frequency of apolipoprotein E ε4 (ApoE ε4) allele in AGD is similar to that of the general population (Tolnay et al., 1998; Togo et al., 2002). Yet the frequency of ApoE ε2 is higher than that observed in AD and controls (Ghebremedin et al., 1998). Polymorphisms of the low-density lipoprotein receptor-related gene protein and α2-macroglobulin gene have also been implicated in AGD (Ghebremedin et al., 2002), paralleling what is known in AD. Nevertheless, association of AGD with tau H1 haplotype is confusing, with both positive and negative results (Togo et al., 2002; Miserez et al., 2003). Further studies are obviously needed to elucidate genetic aspects of AGD.
Clinical symptoms
AGD may manifest with cognitive decline and dementia (Tolnay et al., 2001; Saito et al., 2002; Togo et al., 2002;Tolnay and Clavaguera, 2004). Behavioural abnormalities, personality changes and emotional and mood imbalance have been noted in other cases (Braak and Braak, 1998; Ikeda et al., 2000). Episodic memory loss has been noted in a majority of subjects in some series (Ikeda et al., 2000).
Recent studies have shown that older AGD cases who were admitted to geriatric wards of mental hospitals had suffered from amnesia, irritability and agitation, followed by delusions, dysphoria and apathy (Togo et al., 2005). Mild amnestic cognitive impairment is not uncommon as an initial manifestation of AGD (Jicha et al., 2006; Petersen et al., 2006).
A small number of patients present with progressive transcortical sensory aphasia, prominent abnormal behaviour and aggression, and moderate to severe cognitive impairment consistent with frontotemporal dementia (FTD) (Tsuchiya et al., 2001; Ishihara et al., 2005). These latter examples lend support to the proposal for considering AGD as one of the causes of FTD (Cairns et al., 2007).
Although the variability of lesions and the common accompanying AD pathology make it difficult to assign clinical symptoms to AGs, it is necessary to emphasize the importance of clinical and pathological correlations in individual cases. It is clear that the involvement of the entorhinal cortex, hippocampus, neighbouring temporal cortex and amygdala may be the anatomical substrate for cognitive impairment and dementia in AGD. Using a logistic regression model, AGD has a significant effect on the development of dementia; demented AGD cases show lower stages of AD-related pathology than do pure AD cases, but higher stages than non-demented AGD patients (Thal et al., 2005). Based on these and similar findings, it has been suggested that AGD acts as an additive pathology (Thal et al., 2005; Josephs et al., 2006). It is the presence of AGD plus mild-moderate AD-type pathology that results in dementia and not just the presence of AGD.
Although AGD may present as FTD, this is a very rare situation related with diffuse neocortical involvement (Hodges et al., 2004). Most often, disorders of behaviour and mood imbalance and personality changes are symptoms accompanying AGD in very aged people.
There is no specific test for a clinical diagnosis of AGD. The involvement of structures similar to those affected in patients with mild cognitive impairment and AD makes it difficult to distinguish AD from AGD on the basis of neuroradiological data. Neuroimaging studies have shown atrophy of the anterior part of the temporal and frontal lobes or non-specific cerebral atrophy (Ferrer et al., 2003; Ishihara et al., 2005; Rippon et al., 2005). Moreover, AGD and AD may occur in the same individual. Biological assays do not permit a clinical diagnosis of AGD.
Diagnosis follows neuropathological study of the post-mortem brain. Sometimes, as happened in two personal individual cases with a four-year history of personality changes, mood disorders and mild mental impairment leading to a fulminating disease with hallucinations, ataxia, dementia and sharp EEG complexes, the combination of AGD and CJD was not suspected during life.
Finally, AGD has been reported in 30% of brains from cognitive normal aged (aged 85.4 ± 5.4 years) individuals (Knopman et al., 2003).
Neuropathology of AGD
According to seminal studies, AGs localize in transentorhinal and entorhinal cortex, CA1 area of the hippocampus, presubiculum, neighbouring temporal cortex, orbitofrontal cortex, insular cortex, basolateral nuclei of the amygdala and hypothalamic lateral tuberal nucleus (Braak and Braak, 1989; Schultz et al., 1998). Coiled bodies in oligodendrocytes are common additional findings (Braak and Braak, 1989) (Fig. 1).
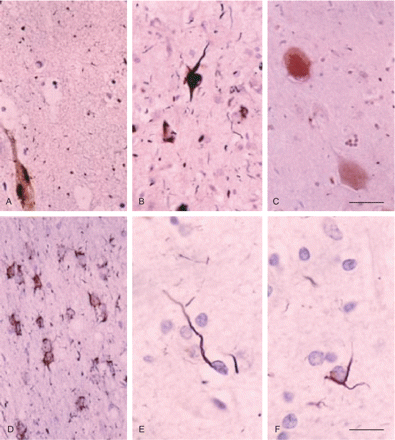
AGD, Gallyas silver staining, showing argyrophilic tangles (A, B), grains (A, B), neuropil threads (B) in the CA1 region (A) and entorhinal cortex (B), periventricular astrocytes (D), and oligodendroglial coiled bodies in the white matter of the temporal lobe (E, F). Ballooned neurons in the amygdala show a diffuse pale coloration (Gallyas negative). Sections lightly counterstained with haematoxylin. A–D, bar in C = 25 microns. E, F, bar in F = 10 microns.
Electron microscopy
AGs contain straight filaments or tubules measuring 9–25 nm (Braak and Braak, 1989; Itagaki et al., 1989; Ikeda et al., 1995). Similar structures have been obtained in sarkosyl-insoluble fractions in AGD cases (Zhukareva et al., 2002). Coiled bodies are composed of accumulations of fibrils measuring 10–13 nm in diameter (Yamada and McGeer, 1990; Ikeda et al., 1995).
Immunohistochemistry
Early studies were mainly based on silver staining. However, the use of immunohistochemistry has greatly expanded the understanding of AGD pathology. The use of anti-tau antibodies has shown that phospho-tau deposition occurs in AGs, as well as in pre-tangle neurons (Gallyas-negative) and coiled bodies in oligodendrocytes. Most tau-containing astrocytes in the limbic system are Gallyas negative (Tolnay et al., 1997a; Botez et al., 1999; Spillantini et al., 1999; Tolnay and Probst, 1999). Based on these crucial observations, AGD is now considered to be a tauopathy.
Grains
The term ‘argyrophilic grains’ is derived from their strong staining with the Gallyas silver iodide method. However, it is worth noting that not all silver methods permit the visualization of AGs (Uchihara, 2007).
Grains are small, about 4–8 microns, spindle shaped, rod-like, button-like or round bodies in the neuropil. Single and double-labelling immunohistchemistry has suggested that AGs are preferentially localized in dendrites and dendritic branches (Ikeda et al., 1995; Schultz et al., 1998; Tolnay et al., 1998), although association of AGs with axons has also been reported (Tolnay and Clavaguera, 2004). The origin of grains is probably in pre-tangle projection neurons of the transentorhinal and entorhinal cortex, CA1 area of the hippocampus, neurons of the dentate gyrus and hilus, presubiculum, neighbouring temporal cortex, orbitofrontal cortex, insular cortex, basolateral nuclei of the amygdala and hypothalamic lateral tuberal nucleus (Tolnay et al., 1998; Tolnay and Clavaguera, 2004).
Studies with the Golgi Cox (Tolnay et al., 1998) and with the rapid Golgi method in the enthorhinal cortex and hippocampus have permitted a clear visualization of AGs as small protrusions of apical, collateral and basilar dendrites of pyramidal cells (Fig. 2). These protrusions grow in normal-appearing dendrites filled with normal dendritic spines. Dendrites and dendritic spines have normal morphology distally to the protrusion. The surface of AGs is smooth, but often they are covered by a few spines or short thin processes (Fig. 2). Although rows of AGs can be seen in the CA1 area of the hippocampus, such rows have not been identified in Golgi-stained sections. These structures largely differ from varicosities, distal stumps and other dendritic abnormalities, as visualized with the Golgi method, in distinct neurological diseases (Ferrer et al., 1990; Ferrer, 2000). AGs also differ from neuritic sprouts as seen in AD (Scheibel and Tomiyasu, 1978; Ferrer et al., 1983; Probst et al., 1983).
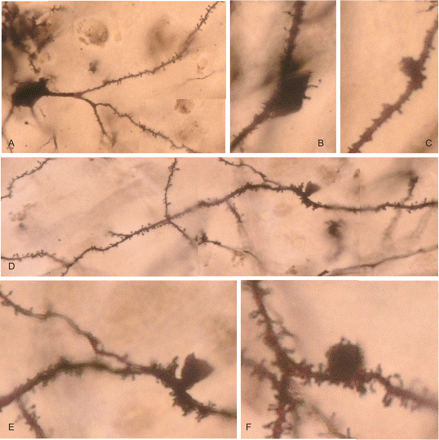
AGD, Rapid Golgi method. (A) Neuron of the entorhinal cortex showing the normal morphology of dendrites and dendritic spines. (B) Spindle-shaped and (C) Round protrusions in dendrites. (D) Basilar dendrite with inverse-cone morphology with normal-appearing proximal (right) and distal (left) processes. (E) High magnification showing the smooth surface of the protrusion and the normal morphology of the spines in the mother dendrite. (F) Round protrusion covered with a few dendritic spines in a normal-appearing dendrite filled with dendritic spines.
Pre-tangle neurons
Pre-tangle neurons are a constant finding in AGD, and their distribution is the same as that for AGs (Tolnay et al., 2003; Tolnay and Clavaguera, 2004). Pre-tangle neurons are immunostained with the same anti-tau phospho-specific antibodies that decorate AGs (Tolnay et al., 1997a; Ferrer et al., 2003). Tau-immunoreactive deposits in granule neurons of the dentate gyrus are a constant abnormality in AGD.
In agreement with previous observations, pre-tangle neurons in AGD do not apparently differ from neurons with early phospho-tau deposition in AD (Braak et al., 1994; Bancher et al., 1989; Tolnay et al., 2003).
Coiled bodies
Coiled bodies are similar to those observed in many other tauopathies and they lack specificity (Chin and Goldman, 1996; Ikeda et al., 1998; Komori, 1999). Yet coiled bodies are a constant finding associated with AGs (Tolnay et al., 2003).
Tau-containing astrocytes
Astrocytes containing hyper-phosphorylated tau show granular immunoreactive cytoplasm rather than dense inclusions like those seen in tufted astrocytes in PSP. Rather, bush-like astrocytes show thin immunoreactive processes with anti-phospho-tau antibodies. Commonly, these delicate processes appear in clusters, thus being reminiscent of thin astrocytic plaques of CBD. In addition to bush-like astrocytes and thin astrocytic plaques in the amygdala, white matter of the temporal lobe, periventricular and subpial astrocytes, tufted astrocytes and small clusters of astrocyte processes reminiscent of astrocytic plaques are also present in some cases.
The presence of tau-containing astrocytes is variable from one case to another. Although usually confined to the limbic system, generalized astrocytosis in the subcortical white matter has been reported in some cases (Yamada et al., 1992; Tsuchiya et al., 2001). Whether these cases are pure AGD or different tauopathies with massive phospho-tau in astrocytes remains unresolved.
Ballooned neurons
Ballooned neurons (Gallyas-negative) expressing αB-crystallin are commonly observed in the amygdala (Tolnay and Probst, 1998), and they have been considered as a marker of AGD (Tolnay and Probst, 1998; Togo and Dickson, 2002). Variable numbers of ballooned neurons are also present in the presubiculum and middle layers of the basal temporal cortex in AGD (Tolnay et al., 2003). Yet ballooned neurons in the limbic system would be more usefully interpreted as a non-specific lesions encountered in many familial and sporadic tauopathies (CBD, PiD, PSP) and AD (Fujino et al., 2004).
The cause of neuron ballooning in AGD and related settings is not known. Ballooned neurons show reduced staining of the rough endoplasmic reticulum. They accumulate phosphorylated neurofilament epitopes, and they are decorated at the periphery with anti-phospho-tau antibodies. Ballooned neurons may be vacuolated. These characteristics show some resemblance to denervated neurons in other regions. The role played by αB-crystallin is also obscure, although recent studies point to its implication in the modulation of cytoskeleton proteins, including filaments and actin, and microtubule assembly (Ghosh et al., 2007a; Gosh et al., 2007b; Ohto-Fujita et al., 2007; Singh et al., 2007).
Tangles and neuropil threads
Variable numbers of tangles and neuropil threads may be present in the same regions as AGs and pre-tangle neurons. This has caused some confusion about the frontier between AGD with a few tangles and AGD with associated AD, and has led to consideration of AGD as a variant of AD (Cras and Perry, 1991). Yet AD changes (neurofibrillary tangles and neuropil threads) in AGD cases can be categorized following the guidelines of Braak and Braak adapted to paraffin sections (Braak and Braak, 1999; Braak et al., 2006).
The instrumental approach with separate consideration of AGs and AD-related changes is suitable, as this convention permits a general commitment for further studies in different settings.
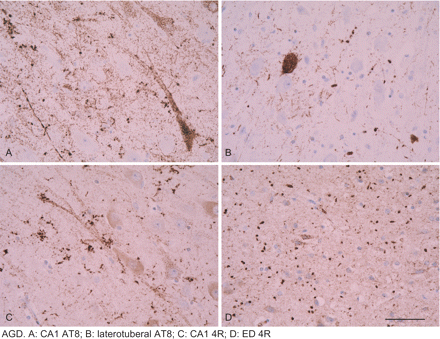
In this line, Braak and collaborators have classified their own AGD cases as AGD + AD 0: 3; AGD + AD I-II: 79; AGD + AD III-IV: 41; AGD + AD V-VI: 2 (Braak and Braak, 1998). The apparently small percentage of AGs in advanced stages of AD must be interpreted cautiously, as the massive phospho-tau-immunoreactive pathology in such cases may hinder the visualization of AGs. This is particularly true when using the monoclonal AT8 antibody. Recent studies using 4R tau-specific antibodies, which highlight AGs, have shown a higher prevalence of AGs in advanced stages of AD (Fujino et al., 2005).
Although often associated with AD, AGD is usually not accompanied by substantial β-amyloid deposits. In contrast to conventional AD cases, AGD brains show a very limited β-amyloid burden in the form of diffuse plaques; neuritic plaques are distinctively uncommon (Tolnay et al., 1999).
Staging of AGs
Early observations indicated that the sole involvement of the anterior part of the CA1 region of the hippocampus was found in apparently normal individuals, whereas involvement of the posterior CA1 region occurred more commonly and more severely in demented cases (Tolnay et al., 1997b). Other studies concluded that severe involvement of the ambient gyrus (the junction between the temporal lobe and the amygdala) differentiated AG with cognitive impairment and dementia from cognitively normal AGD (Saito et al., 2002). More refined analysis has established a proposal for staging AGs (Saito et al., 2004) which basically reflects a similar antero-posterior gradient of putative progression of the disease. Yet rare cases have shown widespread AGs throughout the temporal lobe, limbic system, frontal cortex and brain stem (Tsuchiya et al., 2001; Maurage et al., 2003; Ishihara et al., 2005). The term diffuse AGD has been proposed as a subgroup of AGD to differentiate these cases from the most common limbic AGD (Maurage et al., 2003).
An up-dated staging based on previous descriptions (Braak and Braak, 1998; Saito et al., 2004; Tolnay and Clavaguera, 2004; Ishihara et al., 2005) and personal observations is shown in Table 1.
Argyrophilic grain (AG) staging
| Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|
| Anterior entorhinal cortex; mild involvement of the cortical and basolateral nuclei of the amygdala; mild involvement of the hypothalamic lateral tuberal nucleus | Entorhinal cortex; anterior CA1; transentorhinal cortex; cortical and basolateral nuclei of the amygdala; presubiculum; hypothalamic lateral tuberal nucleus; dentate gyrus | Entorhinal cortex; CA1; perirhinal cortex; presubiculum; amygdala; dentate gyrus; hypothalamic lateral tuberal nucleus; mild involvement of CA2 and CA3; mild involvement of the subiculum; mild involvement of other nuclei of the hypothalamus (i.e. mammillary bodies); mild involvement of the anterior temporal cortex, insular cortex, anterior cingulated gyrus, orbitofrontal cortex, nucleus accumbens, septal nuclei; rare grains in the midbrain | Moderate to severe additional involvement of the neocortex and brainstem |
| Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|
| Anterior entorhinal cortex; mild involvement of the cortical and basolateral nuclei of the amygdala; mild involvement of the hypothalamic lateral tuberal nucleus | Entorhinal cortex; anterior CA1; transentorhinal cortex; cortical and basolateral nuclei of the amygdala; presubiculum; hypothalamic lateral tuberal nucleus; dentate gyrus | Entorhinal cortex; CA1; perirhinal cortex; presubiculum; amygdala; dentate gyrus; hypothalamic lateral tuberal nucleus; mild involvement of CA2 and CA3; mild involvement of the subiculum; mild involvement of other nuclei of the hypothalamus (i.e. mammillary bodies); mild involvement of the anterior temporal cortex, insular cortex, anterior cingulated gyrus, orbitofrontal cortex, nucleus accumbens, septal nuclei; rare grains in the midbrain | Moderate to severe additional involvement of the neocortex and brainstem |
Argyrophilic grain (AG) staging
| Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|
| Anterior entorhinal cortex; mild involvement of the cortical and basolateral nuclei of the amygdala; mild involvement of the hypothalamic lateral tuberal nucleus | Entorhinal cortex; anterior CA1; transentorhinal cortex; cortical and basolateral nuclei of the amygdala; presubiculum; hypothalamic lateral tuberal nucleus; dentate gyrus | Entorhinal cortex; CA1; perirhinal cortex; presubiculum; amygdala; dentate gyrus; hypothalamic lateral tuberal nucleus; mild involvement of CA2 and CA3; mild involvement of the subiculum; mild involvement of other nuclei of the hypothalamus (i.e. mammillary bodies); mild involvement of the anterior temporal cortex, insular cortex, anterior cingulated gyrus, orbitofrontal cortex, nucleus accumbens, septal nuclei; rare grains in the midbrain | Moderate to severe additional involvement of the neocortex and brainstem |
| Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|
| Anterior entorhinal cortex; mild involvement of the cortical and basolateral nuclei of the amygdala; mild involvement of the hypothalamic lateral tuberal nucleus | Entorhinal cortex; anterior CA1; transentorhinal cortex; cortical and basolateral nuclei of the amygdala; presubiculum; hypothalamic lateral tuberal nucleus; dentate gyrus | Entorhinal cortex; CA1; perirhinal cortex; presubiculum; amygdala; dentate gyrus; hypothalamic lateral tuberal nucleus; mild involvement of CA2 and CA3; mild involvement of the subiculum; mild involvement of other nuclei of the hypothalamus (i.e. mammillary bodies); mild involvement of the anterior temporal cortex, insular cortex, anterior cingulated gyrus, orbitofrontal cortex, nucleus accumbens, septal nuclei; rare grains in the midbrain | Moderate to severe additional involvement of the neocortex and brainstem |
Staging of AGs does not include accompanying changes such as pre-tangle neurons, coiled bodies, bush-like astrocytes and ballooned neurons. The number and distribution of pre-tangle neurons parallels the distribution of AGs, whereas coiled bodies and tau-immunoreactive astrocytes have particular patterns in individual cases: large numbers of astrocytes may be encountered with relatively low numbers of AGs. Finally, the number of ballooned neurons in the amygdala shows important individual variation among cases with similar AG stage.
Consideration of the neuropathological diagnosis in AGD cases
Although silver stains have been useful in the past to discover AGD, the Gallyas method and other similar silver stains are not used in current practice. This is due in part to the variations in the quality of staining from one laboratory to another. Moreover, immunohistochemistry has been demonstrated to be a powerful tool with reproducible results in different laboratories (Alafuzoff et al., 2006).
Lesions in AGD are best visualized with the help of any of the several commercial anti-phospho-tau antibodies available; good results are obtained with the AT8 antibody currently used in most laboratories and institutes. The best staining of AGs is achieved with anti-4R tau antibodies (Togo et al., 2002; Fujino et al., 2005). Combining these two antibodies allows the visualization of AGs, neurons with pre-tangles, neurofibrillary tangles, neuropil threads, coiled bodies and different tau-immunoreactive astrocytes.
Ballooned neurons are also mildly phospho-tau-immunoreactive although the best marker of ballooned neurons is anti-αB-crystallin.
The use of additional antibodies (e.g. anti-βA amyloid, α-synuclein, ubiquitin, TDP-43, protease resistant PrP) is obviously necessary to rule out combined pathologies.
Considering the high prevalence of other pathologies in association with AGs, it is probably prudent to consider a double or triple neuropathological diagnosis in the majority of AGD cases (i.e. AG + AD; AG + PSP; AG + CBD + PD). Staging of accompanying diseases may follow international agreement criteria (Braak and Braak, 1999; Braak et al., 2003; McKeith et al., 2005).
The majority of cases may be categorized, for example, as: AGs stage III + AD stage IIIA + PD stage 2, and probably including, as a note, the amount and distribution of tau-immunoreactive astrocytes and the presence of additional details. This complementary note may be useful as it may serve to describe early stages of tauopathies (PSP or CBD) that are commonly associated with AGs and for which there are not, at present, proposals for disease staging like those available for AD and PD.
Biochemistry of tau in AGD
Tau proteins are encoded by the tau gene in chromosome 17. Alternative splicing of exons 2, 3 and 10 results in six isoforms, which in turn give rise to six different mRNAs. The adult tau isoforms are proteins of 441 amino acids (2 + 3 + 10 +), 410 amino acids (2 + 3+ 10 −), 412 amino acids (2 + 3 − 10 +), 381 amino acids (2 + 3 − 10 −) and 383 amino acids (2 − 3 − 10 +); the fetal tau isoform is a protein of 352 amino acids (2 − 3 − 10 −). Tau proteins resulting from encoding exon 10 have four repeat regions (4R tau), whereas those lacking encoding exon 10 have three repeat regions (3R tau) (Goedert et al., 1989; Himmler et al., 1989). The function of tau largely depends on post-translational modifications including phosphorylation and dephosphorylation. Phosphorylation of tau is the result of a balanced action between protein kinases and protein phosphatases. Several kinases have been implicated in tau phosphorylation: glycogen synthase kinase-3 (GSK-3), cyclin dependent kinase-5 (cdk-5), mitogen-activated protein kinase, extracellular signal-regulated kinases (MAPK/ERK1 and MAPK/ERK2, p44 and p42), stress-activated protein kinases, c-Jun N-terminal kinase (SAPK/JNK) and p-38 kinase (p38), among others. These have the capacity to phosphorylate tau at specific sites (Hanger et al., 1992; Mandelkow et al., 1992; Goedert et al., 1997; Lovestone and Reynolds, 1997; Reynolds et al., 1997a; Reynolds et al., 1997b; Goedert et al., 1998; Jenkins et al., 2000; Reynolds et al., 2000; Buée-Scherrer and Goedert, 2002).
AGD is a 4R tauopathy
Early immunohistochemical studies disclosed several sites of tau phosphorylation in AGD (Tolnay et al., 1997a; Ferrer et al., 2003). Sites of tau phosphorylation in AGD do not differ from those in AD. Moreover, in contrast to results from early studies (Tolnay et al., 1997a), tau phosphorylation at Ser262 has been found in AGs and and pre-tangle neurons (Tolnay et al., 2002; Ferrer et al., 2002b). Immunoreactivity of AGs and pre-tangle neurons to non-phosphorylation-dependent antibodies to N-terminal and C-terminal tau suggest that full tau is contained in these structures as well as in coiled bodies (Tolnay et al., 1997a).
Gel electrophoresis of sarkosyl-insoluble fractions has been useful to recognize the band pattern of phospho-tau in AGD. In contrast to AD, characterized by bands of 68, 64 and 60 kDa often accompanied by an upper band of about 73 kDa, AGD is characterized by a double band of 68 and 64 kDa similar to that found in PSP and CBD (Togo et al., 2002; Tolnay et al., 2002; Ferrer et al., 2003). Therefore, AGD is considered a 4R tauopathy (Togo et al., 2002). The use of specific anti-4R antibodies has corroborated this biochemical observation (Togo et al., 2002). Moreover, anti-4R immunohistochemistry has proved a very useful tool for the detection of AGD cases associated with AD changes (Fujino et al., 2005). (Fig. 3).
Strong 4R tau immunoreactivity is observed in pre-tangle neurons, tangles and grains. Paraffin sections, lightly counterstained with haematoxylin. Dilution of the 4R antibody (Upstate) 1:50. Bar = 25 microns.
Interestingly, the presence of tangles and pre-tangles in the hippocampal CA2 area is associated with 4R tauopathy, and the most common is AGD (Ishizawa et al., 2002).
In a particular series, three AGD cases showed a pattern similar to AD, but a 4R profile was found in two other cases (Zhukareva et al., 2002). This example further illustrates the combination of AD and AGD changes in a number of AGD cases.
Truncated forms of tau in AGD
In addition to characteristic bands of 68 and 64 kDa in AGD, bands of lower molecular mass have been illustrated although not described in AGD (Tolnay et al., 2002). This is an important point as bands of low molecular weight are concurrently present in sarkosyl-insoluble fractions in AGD (Fig. 4). These bands are not a post-mortem artefact due to delayed tissue processing (Santpere et al., 2006), but rather they evidence truncated forms of tau (Novak et al., 1991, 1993; Skrabana et al., 2004). The presence of truncated tau may have implications in the pathogenesis of the disease, as truncated tau promotes polymerization of tau in vitro (Abraha et al., 2000), drives neurofibrillary degeneration (Zilka et al., 2006), induces oxidative stress in a rodent model of tauopathy (Cente et al., 2006), and facilitates apoptosis in vitro under appropriate conditions (Fasulo et al., 2000).
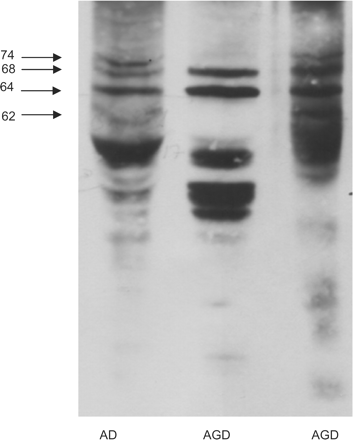
Gel electrophoresis and western blotting of sarkosyl-insoluble fractions of the hippocampus in one case with AD and one pure AGD processed in parallel. Four bands of 74, 68, 64 and 60/62 kDa are characteristic of AD. Two bands of 68 and 64 kDa are seen in AGD. In addition, several bands of lower molecular mass are found in AGD.
Among several proteases capable of cleaving tau, thrombin and prothrombin are present in neurons and accumulate in neurofibrillary tangles in AD (Arai et al., 2005, 2006). By using double-labelling immunohistochemistry and confocal microscopy, co-localization of 4R tau and thrombin is found in AGs (Fig. 5), thus suggesting the participation of thrombin in tau truncation in AGs. In contrast, calpain-2 and active caspase-3 (17 kDa) are only substantially expressed in tangles but not in pre-tangle neurons and AGs in AGD (Fig. 5).
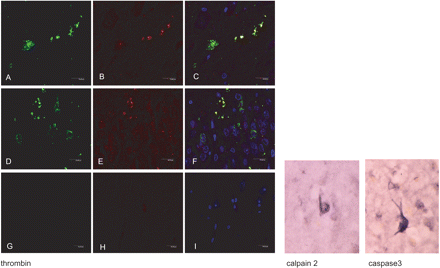
Left panel: Double-labelling immunofluorescence and confocal microscopy showing co-localization of thrombin (green: A, D) and 4R tau (red: B, E) in grains (yellow: C, F) in CA1 area of the hippocampus (A–C) and dentate gyrus (D–F). (G–H): sections incubated without the primary antibodies serve as negative controls. Dilution of the thrombin antibody (American Diagnostica) 1:100 and 4R tau (Upstate) 1:50, respectively. TO-PRO counterstaining (blue) permits the visualization of nuclei. Right panel: calpain 2 (Calbiochem) 1:25 and active caspase 3 (17 kDa) (Cell Signalling) 1:25 immunohistochemistry. Immunoreactivity is restricted to neurofibrillary tangles.
Tau phosphorylation and aggregation in AGD
Pre-tangle neurons contain phospho-tau, and straight filament and tubules, but do not develop paired helical filaments. Similarly, AGs are densely stained with anti-phospho-tau antibodies but they differ from neuropil threads in AD because of this particular structure. The reasons for these differences are barely known and our understanding of the factors that may distinguish pre-tangles and tangles is only fragmentary.
Kinases involved in tau phosphorylation in AGD
A few studies have shown increased expression of active tau-kinases in AGD (Ferrer et al., 2003). These include mitogen activated kinase-extracellular signal-regulated protein kinase 1 and 2 (MAPK/ERK 1 and 2), stress-activated protein kinase (SAPK-JNK), kinase p38, glycogen synthase kinase-3β (GSK-3β) and calcium/calmodulin kinase (CaMK II). Phosphorylated active kinases co-localize with phospho-tau in pre-tangles, AGs, coiled bodies and astrocytes (Ferrer et al., 2003) (Fig. 6). Detailed studies focused on GSK-3β have shown early GSK-3β activation preceding and accompanying the formation of pre-tangles, tangles and other tau-immunoreactive inclusions (Leroy et al., 2007).
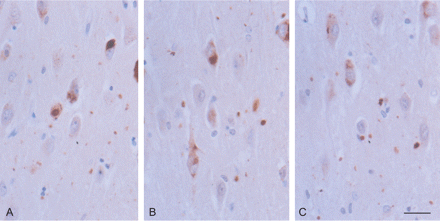
GSK-3 (A), SAPK/JNK-P (B) and p38-P (C) immunoreactivity in the CA1 region of the hippocampus in AGD. Active tau-kinases are expressed in pre-tangle neurons, tangles and grains. Paraffin section lightly counterstained with haematoxylin. Dilution of the antibodies p38-PThr180/Tyr182 (cell Signalling) 1:200; SAPK/JNK-PThr183/Tyr185 (Cell Signalling) 1:150, GSK-3β-PSer9 (Oncogen) 1:150. Bar = 25 microns.
Expression levels of the different tau-kinases in pure AGD (that is, with no associated AD pathology) are similar in total homogenates and sarkosyl-insoluble fractions, a feature which is in contrast with the sequestering of several active kinases in the sarkosyl-insoluble fraction in AD (Ferrer, 2004; Ferrer et al., 2005). Therefore, one of the differential properties of tangles and pre-tangles is the sequestering of tau-kinases in tangles. Immunoprecipitation studies of tau-kinases from paired helical filament-enriched fractions in AD have shown the capacity to phosphorylate specific substrates including recombinant tau, thus indicating that tangles have the ability to recruit tau for further phosphorylation (Ferrer et al., 2002a; Ferrer, 2004; Ferrer et al., 2005).
Components that differentiate pre-tangle neurons from tangles are tubulin (Puig et al., 2005), and elevated levels of iron, ferritin and transferrin in tangles (Quintana et al., 2006).
Sequestosome 1/p62
p62 is a protein for which a role in protein aggregation and degradation has recently been attributed (Seibenhener et al., 2004). p62 is able to self-aggregate and it has high affinity for multi-ubiquitin chains (Vadlamudi et al., 1996; Geetha and Wooten, 2002). Based on this evidence, it has been proposed that p62 may have a role both in promoting protein aggregation and in delivering polyubiquitinated proteins to the proteasome for their degradation (Kuusisto et al., 2001; Nakaso et al., 2004). Previous studies have shown the presence of p62 immunoreactivity in neurons with neurofibrillary tangles and in α-synuclein inclusions within the spectrum of Lewy body diseases (Kuusisto et al., 2002, 2003; Zatloukal et al., 2002). p62 also decorates AGs and to a lesser degree coiled bodies (Scott and Lowe, 2007). Further analysis of p62 expression has shown diffuse moderate p62 immunoreactivity in pre-tangle neurons, sometimes with a peri-nuclear reinforcement, as well as strong localized p62 immunoreactivity in tangles and strong p62 immunoreactivity in grains (Fig. 7A and B). These findings suggest that incorporation of p62 in pre-tangle neurons and grains is probably the same as the mechanism proposed for neurofibrillary tangles in AD (Kuusisto et al., 2002).
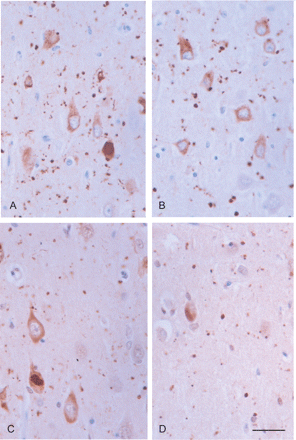
p62 immunoreactivity (A, B) is found in pre-tangle neurons, tangles and grains in the CA1 region of two different AGD cases. Ubiquitin immunoreactivity (C, D) is also observed in pre-tangle neurons, tangles and grains in the same cases. Paraffin section lightly counterstained with haematoxylin. Dilution of anti-p62 C-terminal antibody (Progen) 1:100 and anti-ubiquitin (Dako) 1:500. Bar = 25 microns.
Ubiquitin
The ubiquitin-proteasome system (UPS) plays a crucial role in non-lysosomal protein degradation under certain physiological conditions. Misfolded proteins or un-assembled subunits of larger protein complexes and retro-translocated proteins from the endoplasmic reticulum are also subject to rapid proteasomal degradation (Herschko and Ciechanover, 1998; Glickman and Ciechanover, 2002). The ubiquitin-proteasome pathway is initiated by the conjugation of ubiquitin to the substrate leading to poly-ubiquitilation of the substrate. Most often, the 26S proteasome is composed of two caps or 19S complexes, which are responsible for recognition of ubiquitylated proteins, and the internal barrel 20S catalytic complex with three main peptidase activities: chymotrypsin-like, trypsin-like and peptidylglutamyl peptide hydrolyzing activities (Botchler et al., 1999; Voges et al., 1999; Herschko and Ciechanover, 1998; Glickman and Ciechanover, 2002).
The activity of the UPS is altered in many diseases characterized by aggregation of misfolded or abnormal proteins. Therefore, the proteasome has a crucial secondary role in the pathogenesis of several degenerative disorders including AD, tauopathies and synucleinopathies (Layfield et al., 2003). A common feature of these disorders is the accumulation of ubiquitin bound to non-degraded proteins.
Earlier studies have established that about 50% of AGs and coiled bodies are immunostained with anti-ubiquitin antibodies (Tolnay et al., 2003). However, it is our experience that ubiquitin staining of these lesions is largely sensitive to sub-optimal tissue processing due to long formalin fixation, among other factors. Ubiquitin is present in a majority of AGs and coiled bodies, as well as in many pre-tangle neurons (Fig. 7C and D). Double immunofluorescence and confocal microscopy discloses co-localization of AT8 and ubiquitin in the vast majority of AGs (Fig. 8).
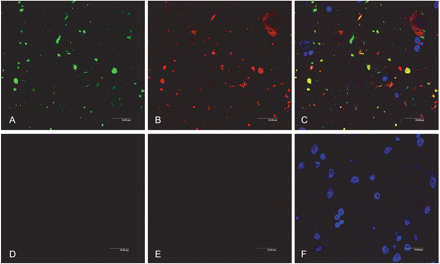
Double-labelling immunofluorescence and confocal microscopy showing co-localization of tau AT8 (green: A) and ubiquitin (red: B) in the majority of grains (yellow: C). G–H: sections incubated without the primary antibodies serve as negative controls. Dilution of AT8 (Innogenetics) 1:50 and ubiquitin (Dako) 1:500. TO-PRO counterstaining (blue) permits the visualization of nuclei.
Mutant ubiquitin (UBB+1)
UBB+1 is generated by a non-DNA-encoded dinucleotide deletion occurring within UBB mRNA. The aberrant protein, resulting from molecular misreading, has a modified C-terminus and is unable to ubiquitinate other protein substrates (van Leeuwen et al., 1998). UBB+1 is itself ubiquitinated, and while at low expression levels it can be degraded by the proteasome, at high levels it can inhibit the proteasomal machinery (Lindsten et al., 2002). Previous studies have demonstrated UBB+1 in neurofibrillary tangles in AD (Fischer et al., 2003). Moreover, UBB+1 accumulates in the hallmark inclusions of Down syndrome and all other tauopathies. Therefore, the accumulation of UBB+1 is considered a specific marker of proteasomal dysfunction in tauopathies and polyglutamine diseases (van Leeuwen et al., 1998; Fisher et al., 2003; de Pril et al., 2004).
Consistent with these previous observations, UBB+1 is expressed in neurons with tangles as well as in AGs in AGD (Fisher et al., 2003). However, very low levels if any are noticed in pre-tangle neurons (Fig. 9). Since the inhibition of the proteasome activity by UBB+1 is dose-dependent (van Tijn et al., 2007), it can be suggested that low levels of UBB+1 in pre-tangle neurons would not impede proteasomal function. High levels of UBB+1 in tangles and grains make these structures barely vulnerable to degradation via the UPS.
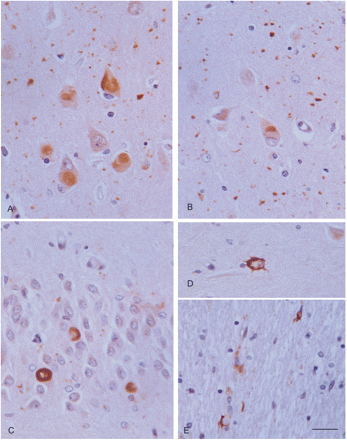
Round elongated deposits of mutant ubiquitin (UBB+1) immunoreactivity in neurons of the CA1 region (A), entorhinal cortex (B), and granule cells of the dentate gyrus (C), as well as in astrocytes (D) and coiled bodies (E). UBB+1 immunoreactivity is clearly present in grains (A, B). Paraffin section lightly counterstained with haematoxylin. Dilution of the rabbit polyclonal UBB+1antibody (Dr Fred W. van Leeuwen, Ubi2+1, 140994, for details see Fisher et al., 2003) 1:400. A, B, bar in B = 25 microns; C–E, bar in E = 10 microns.
It has been shown that paired helical filaments (the main component of neurofibrillary tangles) may further impair the proteasomal function (Keck et al., 2003). It remains to be shown whether grains also have the capacity to collapse the proteasome.
Oxidative stress
Free radical production is a widespread phenomenon occuring under physiological aerobic metabolism in eukaryotic cells, and it is pronounced in the nervous system because of its primordial aerobic metabolism. A balance between oxidation and reduction reactions serves to maintain the physiological redox status. However, oxidative stress may cause deleterious metabolic effects when the generation of reactive oxygen species exceeds the level of antioxidant responses.
Oxidative stress plays a crucial role in the pathogenesis of AD (Moreira et al., 2005; Zhu et al., 2005; Nunomura et al., 2006). One of the consequences of this is the lipoxidation, glycoxidation and nitration of certain proteins (Pamplona et al., 2005; Butterfield et al., 2006a; Butterfield et al., 2006b; Sultana et al., 2006a; Sultana et al., 2006b; Sultana et al., 2006c), which results in loss of function. Decline in the proteasome function in the earliest stages of AD is partially due to oxidative inactivation (Cecarini et al., 2007). Another consequence is the activation of signal transduction cascades including stress kinases (Petersen et al., 2007).
In spite of the important information about oxidative stress in AD, practically nothing is known about its role in AGD. Yet increased expression of advanced glycation end products (AGEs) and AGE receptor (RAGE) can be seen by immunohistochemistry in neurons of the hippocampus and entorhinal cortex in pure forms of AGD (data not shown). AGEs are carbonyl groups generated by secondary reaction of the primary amino group of lysine residues with reactive carbonyl derivatives produced by the reaction of reducing sugars or their oxidation products with lysine residues of proteins (glycation/glycoxidation reactions) (Dalle-Donne et al., 2006). RAGE is a member of the immunoglobulin superfamily sensitive to the generation of reactive oxygen species that is crucial for many AGE-induced changes in cellular properties, including activation of protein kinase pathways (Lander et al., 1997; Schmidt et al., 2000). Anti-oxidant responses can also be determined by immunohistochemistry to Cu/ZN superoxide dismutase I (SOD1) and Mn superoxide dismutase II (SOD2) in the AGD brain (data not shown). Increased SOD1 and SOD2 immunoreactivity is observed in neurons in vulnerable regions in parallel with AGs and pre-tangle neurons. These findings show that increased oxidative stress and increased oxidative responses occur in the hippocampus and entorhinal cortex in pure AGD, and, therefore, that these modifications are unrelated to typical AD changes.
Evidence of oxidative stress also suggests a link between free radical production, stress kinase (basically, SAPK/JNK and p38) activation, and tau phosphorylation in AGD. Similar scenarios have been reported in AD and other human and murine tauopathies (Zhu et al., 2000, 2001, 2003; Puig et al., 2004; Ferrer, 2004, Ferrer et al., 2005).
Neurotransmitters and receptors
Little is known about neurotransmitters and receptors in AGD. A single study reported normal cortical levels of choline acetyltransferase but markedly reduced levels of dopamine and its metabolites in the striatum (Yamada et al., 1992). However, the description of these two cases is consistent with AGD associated with progressive subcortical gliosis.
Recently, increased adenosine receptor A (A1), but not A2A or A2B, together with increased levels of adenylyl ciclase, an effector of A1, and sensitization of this pathway, has been reported in the hippocampus but not in the frontal cortex in pure AGD (Perez-Buira et al., 2007). This has been interpreted as a compensatory response geared to modulating glutamate neurotransmission and facilitating neuroprotection of this preferentially involved region in AGD.
Concluding comments
Although very common, AGD is still a poorly understood neurological disorder. However, the recent studies discussed above allow us to delineate a pathogenic scenario for some hallmark aspects of the disease. A summary of the pathways involved and suggested activation of metabolic cascades is shown in Fig. 10. Aging is the most important determinant factor. Oxidative stress plays a crucial role in the activation of stress-activated tau-kinases, which facilitate tau-hyperphosphorylation in a subset of neurons. Abnormal tau is aggregated after binding with p62 and subsequently ubiquitinated in grains, pre-tangle neurons and tangles. Incorporation of mutant ubiquitin, UBB+1, blocks hyperphosphorylated tau degradation in grains and tangles. These structures sequester active tau-kinases, thus further promoting local tau hyperphosphorylation in AGs and tangles. Finally, thrombin accumulated in AGs and tangles may facilitate tau truncation, which is toxic for cells and which increases oxidative stress.
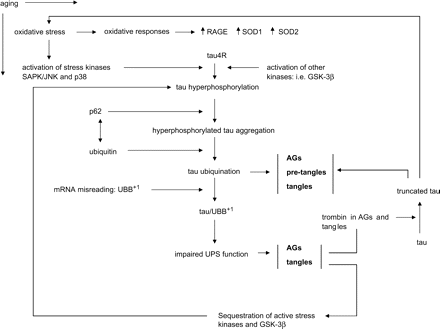
Summary of pathologic events in AGD.
In spite of these achievements, several points remain obscure, such as: (i) original causes leading to oxidative stress; (ii) targets of oxidative stress; (iii) role played by phosphatases; and (iv) reasons for selective tau 4R hyperphosphorylation, among others.
AGD is restricted to human beings, as no similar lesions have been reported under natural conditions in animals. Yet deafferentation of the hippocampus after lesions of the entorhinal cortex in rats is followed by the presence of small granules containing phospho-tau in the molecular layer of the dentate gyrus (Mudher et al., 2001). Whether combined deafferentation and abnormal tau metabolism localized in vulnerable points of the dendritic arbour may be causative of grain architecture is a fascinating working hypothesis.
Acknowledgements
This study was supported in part by the Spanish Ministry of Health, Instituto de Salud Carlos III (PI05/1570 grant) and CIBERNED program, the European Commission, under the Sixth Framework Programme (BrainNet Europe II, LSHM-CT-2004-503039) and ISAO grant 06502. Brain samples were obtained from the Institute of Neuropathology and University of Barcelona Brain Banks following the guidelines and approval of the local ethics committees. We wish to thank Dr Jesús Ávila, Centro de Biología Molecular Severo Ochoa, Universidad Autónoma de Madrid, for providing the tau antibody 7.51; Rosa Blanco, Margarita Carmona (Institut de Neuropatologia) and Benjamín Torrejón-Escribano (Serveis Científico-Tècnics, Unitat de Biología de Bellvitge) for technical assistance; and Tom Yohannan for editorial help.
References
Abbreviations:
- AD
Alzheimer's disease
- AGD
argyrophilic grain disease
- AG
argyrophilic grains
- CBD
corticobasal degeneration
- CJD
Creutzfeldt–Jakob disease
- DLB
dementia with Lewy bodies
- FTD
frontotemporal dementia
- PD
Parkinson's disease
- PiD
Pick's disease
- PSP
progressive supranuclear palsy
- RAGE
AGE receptor
- UPS
ubiquitin-proteasome systemb


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}