




Abstract
B cells are increasingly recognized as major players in multiple sclerosis pathogenesis. The BAFF/APRIL system is crucial for B cell homoeostasis and may drive B cell-dependent autoimmunity. We asked whether this system is affected by Interferon (IFN)-β therapy. We analysed transcription of the ligands (BAFF, APRIL, TWE-PRIL) and the corresponding receptors (BAFF-R, TACI and BCMA) by TaqMan-PCR ex vivo in whole blood and in immune cell subsets purified from IFN-β-treated multiple sclerosis patients. Serum BAFF concentrations were determined by ELISA. This cross-sectional study involved 107 donors. IFN-β therapy strongly induced BAFF transcription proportionally to the IFN-β biomarker MxA in monocytes and granulocytes in vivo. BAFF serum concentrations were elevated in IFN-β-treated multiple sclerosis patients to a similar level as observed in SLE patients. In cultured PBMC, neutrophils, fibroblasts and astrocytes, BAFF was induced by IFN-β concentrations similar to those reached in vivo in treated multiple sclerosis patients. BAFF turned out to be the main regulated element of the BAFF/APRIL system. In untreated multiple sclerosis patients, there was no BAFF increase as compared to healthy controls. Our study reveals a complex situation. We show that IFN-β therapy induces a potent B cell survival factor, BAFF. However, B cell depletion would be desirable at least in some multiple sclerosis patients. The systemic induction of BAFF by IFN-β therapy may facilitate the production of various autoantibodies and of IFN-neutralizing antibodies. Individual MS/NMO patients who have major B cell involvement may benefit less than others from IFN-β therapy, thus explaining interindividual differences of the therapeutic response.
Introduction
B cells and antibodies are increasingly recognized as important elements in the pathogenesis of multiple sclerosis. Accordingly, the first B cell targeting therapies, such as rituximab and atacicept, are currently under investigation (Hemmer and Hartung, 2007; Hauser et al., 2008). One of the most crucial factors for the maintenance of B cells is the BAFF (B cell activating factor of the TNF family)/APRIL (a proliferation-inducing ligand) system. It regulates B cell survival, differentiation and class switching, and determines the size of the peripheral B cell pool (Schneider, 2005; Mackay et al., 2007). This system comprises three ligands (BAFF, APRIL, TWE-PRIL), and three signalling receptors (BAFF-R, TACI, BCMA.). Current evidence indicates that BAFF (TNFSF13b, BLyS) is the most important of the three ligands (Schneider, 2005; Mackay et al., 2007).
BAFF is induced at sites of inflammation, suggesting that BAFF can exacerbate and perpetuate local inflammation by stimulating the survival of B cells in inflamed tissues (Tan et al., 2003; Krumbholz et al., 2005a; Tschen et al., 2006). Specifically, we previously reported that BAFF is induced in astrocytes in multiple sclerosis lesions, supporting its role as a B cell fostering factor acting in the local milieu of multiple sclerosis lesions (Krumbholz et al., 2005a). Furthermore, elevated systemic levels of BAFF and/or APRIL have been observed in serum of patients with systemic, B cell-related autoimmune diseases such as Sjögren's syndrome (Groom et al., 2002; Jonsson et al., 2005), systemic lupus erythematosus (SLE) (Stohl et al., 2003; Koyama et al., 2005), and Wegener's granulomatosis (Krumbholz et al., 2005b). In patients infected with hepatitis C virus (HCV), elevated serum BAFF levels were associated with clinical and laboratory features of autoimmunity (Toubi et al., 2006). Functional evidence for a causal link between high BAFF levels and B cell-dependent autoimmunity has been obtained in animal models: BAFF-overexpressing mice develop B cell hyperplasia, elevated immunoglobulin (Ig) levels, autoantibodies and clinical autoimmune diseases (MacKay et al., 1999; Kalled 2005). Autoreactive B cells seem to be especially dependent on BAFF for survival, and may gain selective advantage when increased amounts of BAFF are available (Lesley et al., 2004).
Interferon-β (IFN-β) is a standard treatment for relapsing-remitting multiple sclerosis. Together with IFN-α and IFN-ω, IFN-β belongs to the group of type I interferons (IFN-I). These are naturally produced cytokines, which play an important role in the defence against infections. Under pathological conditions, notably SLE, high systemic levels of naturally produced IFN-α are thought to play a central pathogenic role (Banchereau and Pascual, 2006). Therapeutic administration of IFN-I can also be accompanied by development of autoantibodies and appearance of clinical autoimmunity: IFN-α is known to induce autoantibodies in 4 to 19% of treated patients (Banchereau and Pascual, 2006). Symptomatic autoimmune reactions, e.g. affecting the thyroid gland in 0.6–7% (Borg and Isenberg, 2007), have also been described as a complication of treatment with IFN-α. Although to a lesser extent, induction of autoantibodies, and in few cases, symptomatic autoimmunity, have also been observed during therapy with IFN-β (Blake and Murphy, 1997; Durelli et al., 1999; Bitsch et al., 2004; Dionisiotis et al., 2004; Alanoglu et al., 2007; Borg and Isenberg, 2007). Viewed together, all these observations suggest that IFN-I can promote humoral immunity, either directly (Le Bon et al., 2006) or indirectly (Le Bon et al., 2001).
In the present study, we addressed the question whether and how treatment with IFN-β affects the BAFF/APRIL system in multiple sclerosis. Our results demonstrate that especially BAFF is strongly upregulated in the majority of patients during IFN-β therapy. This observation offers a plausible link between IFN-I, BAFF, and B cell-related autoimmunity, opening new perspectives for improving therapy.
Patients, Materials and Methods
Patients and healthy blood donors
We obtained peripheral blood samples from 73 multiple sclerosis patients (50 women, 23 men, median age 35 years). Forty-seven patients were treated with one of the available IFN-β preparations [11 patients with Avonex (6 MIU IFN-β1a 1 × /week; Biogen Idec, Cambridge, MA, USA), 29 with Rebif 11–44 µg (3–12 MIU IFN-β1a 3 × /week; Serono, Geneva, Switzerland) and seven with Betaferon (8 MIU IFN-β1b every 2 days; Schering, Berlin, Germany)]. Twenty-six patients were untreated at the time of sampling. From the IFN-β-treated patients blood was drawn 12–24 h after the last injection, an appropriate time window for showing IFN-β-induced transcriptional changes (Hoffmann et al., 2007).
Nineteen of the IFN-β-treated patients had a concomitant medication of non-steroidal anti-inflammatory drugs (paracetamol/acetaminophen or ibuprofen) for symptomatic treatment of IFN-β-related adverse effects. One IFN-β-treated patient had an additional short course of i.v. corticosteroids 1 week before blood sampling. No other immunomodulatory or immunosuppressant medications were taken during 4 weeks prior to sampling.
In addition, we obtained 12 serum samples from eight IFN-α-treated melanoma patients (one woman, seven men, median age 49 years). Patients 1–4 received a high dose of IFN-α (20 MIU/d i.v.), and patients 5–8 a lower dose of IFN-α (3 × 3.3 MIU/week s.c.). From Patients 1–3, one sample was obtained before and one during high-dose IFN-α therapy. Two samples from Patient 4 were obtained during high-dose therapy. The samples from Patients 5–8 were obtained 1 day after injection.
Samples from 26 healthy donors (median age 31 years) served as negative controls. The number of samples used in each assay is indicated by showing individual data points in the figures. This study was reviewed and approved by the Ethics Committee of the Ludwig-Maximilians-University of Munich. Informed consent was obtained according to the Declaration of Helsinki.
Cell separation and cell culture
Peripheral blood mononuclear cells (PBMC) were isolated from fresh EDTA-treated blood by density gradient centrifugation. Monocytes were isolated from PBMC using anti-CD14 labelled magnetic microbeads (MACS, Miltenyi, Bergisch Gladbach, Germany). Neutrophils were isolated from the pellet of the density gradient by four cycles of hypotonic lysis of erythrocytes (ice-cold 0.2% NaCl for 30 s, reconstitution of isotonicity with 1.6% NaCl). Human astrocytes of embryonic origin (Krumbholz et al., 2005a) were used after the third or fourth passage. Owing to their embryonic origin, these astrocyte cultures are devoid of microglial cells or macrophages (Krumbholz et al., 2005a; Aloisi et al., 1992).
All cells were cultured in RPMI supplemented with 5–10% FCS (PAN Biotech, Aidenbach, and Biochrom, Berlin, Germany). Cells were stimulated overnight with IFN-β1b (Betaferon, Schering, Berlin, Germany) or IFN-γ (Roche, Diagnostics GmbH, Mannheim, Germany).
RNA and cDNA preparation, quantitative PCR
RNA from isolated or cultured cells was prepared using RNeasy Mini columns including an on-column DNase digestion step. Whole-blood RNA was obtained using the QIAamp RNA Blood Mini Kit or the PreAnalytix PAXgene Blood RNA collection system in conjunction with the PAXgene Blood RNA Kit (all Qiagen, Hilden, Germany).
RNA from blood was analysed from two sets of donors enrolled in different years, each including healthy controls, untreated, and IFN-β-treated multiple sclerosis patients. For the first set, RNA was transcribed using the High Capacity cDNA Kit with random hexamer primers, and analysed on TaqMan low-density arrays on an ABI 7900 using Universal Mastermix (all ABI, Darmstadt, Germany). For the second set and for cultured cells, RNA was transcribed using M-MLV (Promega, Mannheim, Germany) and random hexamer primers, and analysed by quantitative PCR in the 96-well format on the ABI 5700 using the qPCR Core kit (both Eurogentec, Seraing, Belgium). Differences in the spectral sensitivity for the fluorescence signal between these two Taqman PCR machines were normalized according to the median expression levels of healthy controls and non-treated multiple sclerosis patients in these two sets of donors. This was validated by essentially the same fold-induction of BAFF and MxA in both sets of donors as described in results. BAFF (Krumbholz et al., 2005a), MxA, TWE-PRIL and APRIL primers and probes (Supplementary Table 1) were designed to be intron-spanning, not amplifying genomic DNA. Efficiency of amplification for qPCR was validated according to ABI instructions.
ELISA
ELISAs were performed as described (Krumbholz et al., 2005a) using anti-human BAFF mAb clone B4H7.2 (5 µg/ml) for coating, and biotin-labelled anti-human BAFF clone A9C9.1 (1 µg/ml) for detection (Biogen-Idec, Cambridge, MA, USA).
Results
IFN-β therapy stimulates BAFF transcription in vivo
We analysed blood RNA samples from 20 healthy donors and 61 multiple sclerosis patients, 39 of whom were treated with one of the three IFN-β preparations approved for multiple sclerosis therapy. Treated patients were classified as biological MxA responders (n = 29) or MxA non-responders (n = 10) based on the induction of the IFN-β response biomarker MxA (Bertolotto et al., 2003; Pachner et al., 2003; Hoffmann et al., 2007). We quantified BAFF expression by TaqMan PCR: Untreated multiple sclerosis patients and IFN-β-treated MxA non-responders had similar levels of BAFF transcripts in blood cells, which were also similar to healthy controls (Fig. 1A). However, IFN-β-treated MxA responders showed a marked induction of BAFF (P < 0.01, Fig. 1A). BAFF expression correlated with the bioavailability of IFN-β as measured by the expression of MxA (Spearman's test: R = 0.85, P < 0.001). These data were derived from two sets of donors (52 and 29 subjects, respectively), each including healthy controls, non-treated, IFN-β-treated MxA non-responding and responding multiple sclerosis patients. A similar induction of BAFF was seen in both sets of patients: the up-regulation of BAFF was 5.0- and 5.5-fold and that of MxA 24.2- and 24.0-fold for the first and second set of patients, respectively.
![IFN-β therapy induces BAFF transcription in multiple sclerosis patients. Whole blood samples were collected from IFN-β-treated multiple sclerosis patients 12–24 h post-injection, untreated multiple sclerosis patients (NT) and healthy blood donors (HC). BAFF (A) and MxA [(an indicator for IFN-β bioactivity, (B)] transcripts were quantified by TaqMan PCR. While there was no difference between healthy controls and untreated multiple sclerosis patients, BAFF transcription was induced in IFN-β-treated multiple sclerosis patients who were biological responders (R) according to a positive MxA induction, but not in non-responders (NR). In contrast, there was only a slight increase of APRIL (C) in the IFN-β-treated MxA responding multiple sclerosis patients compared to untreated patients (ANOVA on ranks/Dunn's test, **: P < 0.01).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/131/6/10.1093_brain_awn077/1/m_awn077f1.gif?Expires=1716354899&Signature=ltgj8-2SBv9u5-kHGAgvVxJ85mrGSFAQUodoftgRFQjlS-ClAGXsNEIEuoCbkO1OG1h4APP6UTJoDYIfuLK8geupi3eRkQKhSulQbk8ljROrUBPB3LNGaEO4BrgruU-m2z7~rX9Q-areRRGst~pbm~-xMhdxOlpx8HZBKExdMMDpSFclVxVmVs98WrPQO-RsFUJMs4tQMFxl98CQ-RwBRyNvSVmZN9qal3zSj68wuVtpMCain8QacUn34tvYg6sgwdpga2UCKJVMNNmi3pMshFNGguXJ436uy39BKrM4rLMSkEyAPdkpgZwgfLnAn2wjtDkVz9ez2Qo~Ksae1VfeFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IFN-β therapy induces BAFF transcription in multiple sclerosis patients. Whole blood samples were collected from IFN-β-treated multiple sclerosis patients 12–24 h post-injection, untreated multiple sclerosis patients (NT) and healthy blood donors (HC). BAFF (A) and MxA [(an indicator for IFN-β bioactivity, (B)] transcripts were quantified by TaqMan PCR. While there was no difference between healthy controls and untreated multiple sclerosis patients, BAFF transcription was induced in IFN-β-treated multiple sclerosis patients who were biological responders (R) according to a positive MxA induction, but not in non-responders (NR). In contrast, there was only a slight increase of APRIL (C) in the IFN-β-treated MxA responding multiple sclerosis patients compared to untreated patients (ANOVA on ranks/Dunn's test, **: P < 0.01).
We further dissected BAFF induction by analysing different immune cell subsets isolated directly from IFN-β-treated patients. IFN-β therapy induced BAFF proportionately to MxA in monocytes (Spearman's test: R = 0.78, P < 0.0001) and granulocytes (R = 0.965, P < 0.0001), but not in lymphocytes (Fig. 2). In order to address the kinetics of BAFF transcription in vivo, we analysed serial blood samples from patients treated with the three approved IFN-β preparations 12–48 h post-injection. Transcription of both BAFF and the IFN-β response biomarker MxA was maximal ≤12 h post-injection. Both genes declined simultaneously and slowly over the next few days, but were still clearly upregulated 24 h post-injection (data not shown).
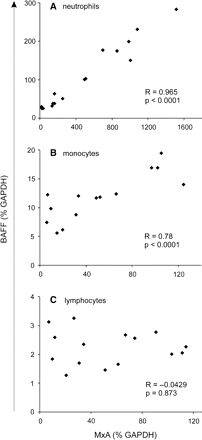
BAFF induction by IFN-β therapy in monocytes and granulocytes. White blood cell subsets from IFN-β-treated multiple sclerosis patients were isolated and analysed by TaqMan PCR. The IFN-β bioactivity marker MxA was induced in all cell types. BAFF expression correlated with MxA in cells capable of synthesizing BAFF (monocytes and granulocytes), but not in lymphocytes (Spearman's test).
IFN-I therapy is accompanied by elevated BAFF serum concentrations
The serum BAFF concentrations of untreated multiple sclerosis patients were similar to healthy controls (Fig. 3A). In multiple sclerosis patients who showed a biological response (MxA induction) to IFN-β therapy (6 MIU 1×/week to 12 MIU 3×/week i.m. or s.c.), the serum BAFF concentrations were elevated ∼1.5-fold (P < 0.01, ANOVA on ranks/Dunn's test, Fig. 3A). This was comparable to melanoma patients treated with IFN-α 3 × 3 MIU/week s.c., whereas melanoma patients receiving ultra-high-dose IFN-α therapy (20 MIU/d i.v.) had even higher BAFF serum levels (Fig. 3A). In all three melanoma patients from whom serum was available before and during IFN-α therapy, there was a clear increase of the BAFF serum concentration (Fig. 3B).
![IFN-I therapy increases systemic BAFF availability. (A) BAFF serum concentrations were determined by ELISA in healthy controls (HC), untreated multiple sclerosis patients (NT-MS), IFN-β-treated MxA responding multiple sclerosis patients (IFN-β) and IFN-α-treated melanoma patients [IFN-α; high dose (10–20 MIU/d i.v.): closed triangles, lower dose (3 × 3.3 MIU/week s.c.): open triangles)]. Whereas NT-MS patients had essentially the same serum levels as healthy controls, we observed a moderate BAFF serum elevation in the IFN-β-treated MxA responders, and a marked elevation especially in the high-dose IFN-α-treated melanoma patients. The medians are indicated for each group. (ANOVA on ranks/Dunn's test, **: P < 0.01, n.s.: not significant). (B) In three melanoma patients, serum samples were available before initiation of and during IFN-α therapy, which led to a clear intra-individual increase in BAFF serum concentrations.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/131/6/10.1093_brain_awn077/1/m_awn077f3.gif?Expires=1716354899&Signature=ZfDm54oKo0MU-eynMxFzxZVu-FXbkFtHupkM8-uXsoA1CFhF7MLKNC-AQ27I784muNNH-kYFap6iRfy43UGXmUJtyBwcPG-ytLbAQFMFcySlvUKMWfRByLG2Sh~nztJTIwzacrJyV1GOCG9mc8muggOaWCYgl6C-blKw-vuJ1TFhrqgYKB482k0NGV4x18HyCr4sf6Bf4CkMcmFjDzqNDPqrOZ~VZlBYbNT9ZJq5EQByiQArm-O2-mJ6M48uCAM5fqBwVZ6iSTVSSICbprJKqyyL3ED27f8n4vl7NM4Cb8Po3vBRvzycarIxn7Nhv-zlrIzCfI4~1ZAd2ljvBykiNg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IFN-I therapy increases systemic BAFF availability. (A) BAFF serum concentrations were determined by ELISA in healthy controls (HC), untreated multiple sclerosis patients (NT-MS), IFN-β-treated MxA responding multiple sclerosis patients (IFN-β) and IFN-α-treated melanoma patients [IFN-α; high dose (10–20 MIU/d i.v.): closed triangles, lower dose (3 × 3.3 MIU/week s.c.): open triangles)]. Whereas NT-MS patients had essentially the same serum levels as healthy controls, we observed a moderate BAFF serum elevation in the IFN-β-treated MxA responders, and a marked elevation especially in the high-dose IFN-α-treated melanoma patients. The medians are indicated for each group. (ANOVA on ranks/Dunn's test, **: P < 0.01, n.s.: not significant). (B) In three melanoma patients, serum samples were available before initiation of and during IFN-α therapy, which led to a clear intra-individual increase in BAFF serum concentrations.
Some individuals have excessively high BAFF levels
We previously reported that a few healthy blood donors who had no clinical signs or symptoms, nor any relevant laboratory abnormalities, show unexplained, excessively high serum levels of BAFF, which were stable over time (Krumbholz et al., 2005b). Here, we identified two additional subjects with >60 ng/ml BAFF in the serum. Thus the frequency is within the same order of magnitude as previously described (Krumbholz et al., 2005b) (2–6% of all subjects). We could obtain whole blood for PCR analysis from four of these individuals [two healthy blood donors, one untreated multiple sclerosis patient, one IFN-β-treated multiple sclerosis patient (MxA responder)]. They all had normal BAFF mRNA expression (1.3-, 0.6-, 0.8- and 0.6-fold of the median of the respective donor group), pointing to an additional cell-specific, post-transcriptional, or proteolytical pathway of BAFF regulation in these particular subjects; further studies will have to elucidate the background of this observation.
IFN-β is a stronger inducer of BAFF than IFN-γ in cultured stromal and immune cells
We extended our study to investigate the in vitro effects of IFN-β on several relevant cell types. IFN-β induced marked BAFF expression in PBMC, neutrophils, fibroblasts (Fig. 4) and astrocytes (Fig. 5). Compared with the known BAFF-inducing cytokine IFN-γ, IFN-β had a stronger effect: In dose–response experiments, IFN-β stimulation resulted in a higher maximum transcription than was observed for saturating amounts of IFN-γ in neutrophils, PBMC, and fibroblasts (Fig. 4). Among these cell types, neutrophils responded to the lowest IFN-β concentrations and showed the highest BAFF transcription relative to the housekeeping gene.
![IFN-β is a stronger inducer of BAFF than IFN-γ in neutrophils (A), peripheral blood mononuclear cells [(PBMC, (B)], and fibroblasts [(HFF, (C)]. Cells were incubated with increasing concentrations of IFN-β and IFN-γ overnight. BAFF expression was measured by TaqMan PCR. IFN-β induced BAFF expression in all cell types. The maximal inducible expression was higher with IFN-β than with IFN-γ. In contrast, APRIL and TWE-PRIL were barely or not at all induced. Due to low expression, not all data points for TWE-PRIL are visible. The legend shown in (A) is valid for all cell types. Error bars indicate SD of replicates.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/131/6/10.1093_brain_awn077/1/m_awn077f4.gif?Expires=1716354899&Signature=yzTlSdo-9wdqoND9eHiAhh3nE2hkmYjUQiCAfW1BL2a1fTzv6aDOKZwqr2BSJRqDHUxXtEXrXthtDYZbKyxjAlAv0t9Z3sQKYWrq1dz9teEbBgB9VqegyVbXtgVj8XMF59MagiM9srD5EHU3KsDGaeqUAonkRRIhVMF-yirMuw5iJ9-09J03yMhC1S2zRZEy7dP7Ck2Omz9k4ZaxvaghQMjGUpi2z8kjrC21a~w-paht9jhKcTXxScIU0FPh2m513j3fxxlKJEDdYOvYw6IN038QM9FvG1l1Z933XC2FbXRF0uMNigzPQFdav2xlwBFkq0jsDadU6OMQQqt~YZDYfA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IFN-β is a stronger inducer of BAFF than IFN-γ in neutrophils (A), peripheral blood mononuclear cells [(PBMC, (B)], and fibroblasts [(HFF, (C)]. Cells were incubated with increasing concentrations of IFN-β and IFN-γ overnight. BAFF expression was measured by TaqMan PCR. IFN-β induced BAFF expression in all cell types. The maximal inducible expression was higher with IFN-β than with IFN-γ. In contrast, APRIL and TWE-PRIL were barely or not at all induced. Due to low expression, not all data points for TWE-PRIL are visible. The legend shown in (A) is valid for all cell types. Error bars indicate SD of replicates.
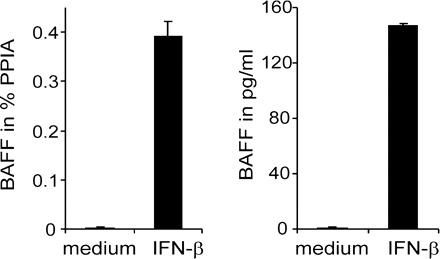
IFN-β induces BAFF in astrocytes. Astrocytes were cultured in the presence or absence of IFN-β. BAFF transcription and secretion into the supernatant was measured by TaqMan PCR and ELISA. IFN-β induced both BAFF mRNA expression and protein secretion. Error bars indicate SD of replicates.
BAFF, APRIL and TWE-PRIL are differentially regulated
APRIL and BAFF were expressed at the same order of magnitude in peripheral blood from healthy donors and untreated multiple sclerosis patients in vivo (Fig. 1A and C), whereas TWE-PRIL was expressed about ≥1 order of magnitude less (data not shown). The same pattern was evident in secondary lymphatic tissue (tonsils, data not shown). Thus we confirm the existence of the hybrid transcript TWE-PRIL in vivo, but due to its low abundance it is presumably of less biological importance than BAFF or APRIL.
After stimulation with IFN-β and other inflammatory cytokines in vitro, regulation of both APRIL and TWE-PRIL was minor compared with the strong induction of BAFF in all analysed cell types (PBMC, monocytes, granulocytes, fibroblasts, astrocytes) (Fig. 4 and data not shown). Likewise, IFN-β therapy did not induce TWE-PRIL in vivo (factor 0.995 in 4 IFN-β-treated MxA responders). APRIL expression was also only slightly, but significantly, upregulated in leucocytes from multiple sclerosis patients who responded to IFN-β therapy by MxA transcription when compared to untreated patients (ANOVA on ranks/Dunn's test, P < 0.01, Fig. 1C). APRIL expression correlated with MxA as a marker for IFN-I bioavailability (R = 0.66, P < 0.001, Spearman's test).
Transcription of BAFF/APRIL receptors is not detectably regulated by IFN-β therapy
The three receptors of the BAFF/APRIL system, BAFF-R, BCMA, TACI could be detected in peripheral blood from all subjects independent of IFN-β treatment (Fig. 6). This was consistent with in vitro experiments, where IFN-β also had no substantial effect on receptor expression in PBMC and granulocytes (data not shown).
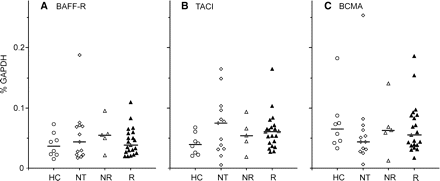
BAFF receptor expression is not influenced by IFN-β in vivo. Whole blood samples were analysed by TaqMan PCR for the expression of the BAFF receptors in healthy controls (HC), untreated multiple sclerosis patients (NT), IFN-β-treated MxA responders (R) and non-responders (NR). Transcription of BAFF-R, TACI and BCMA was not changed significantly by IFN-β therapy.
Discussion
We show that in multiple sclerosis, treatment with IFN-β leads to strong upregulation of BAFF in blood leucocytes and serum. The results provide a link between the known propensity of type I IFN to stimulate humoral (auto-) immunity, and the established role of BAFF as a crucial survival factor for B cells.
Compared to healthy controls and untreated multiple sclerosis patients, BAFF serum concentrations were elevated ∼1.5-fold, which is in a similar range as observed in the systemic B cell-related autoimmune disease SLE [(1.2- to 2.5-fold upregulation (Cheema et al., 2001; Zhang et al., 2001; Groom et al., 2002)], where IFN-α is thought to be a major pathogenic factor (Banchereau and Pascual, 2006). BAFF transcription in blood cells (monocytes and neutrophils) was induced around 5-fold in IFN-β-treated patients. The difference of BAFF induction between the transcript and protein levels might be explained by the observation that enhanced availability of BAFF leads to enhanced consumption and absorption of BAFF via BAFF-R on B cells (Carter et al., 2005), consistent with the view that BAFF is indeed one of the limiting factors determining the size of the peripheral B cell pool (Schneider, 2005).
Elevated levels of BAFF facilitate and foster B cell-mediated immune reactions: As shown in animal models, elevations of BAFF favour B cell-dependent autoimmune reactions (MacKay et al., 1999). Furthermore, autoreactive B cells have an increased demand for BAFF for survival, and may be selectively expanded when increased amounts of BAFF are produced (Lesley et al., 2004). Therefore, the elevation of BAFF by IFN-β we describe here could plausibly explain why IFN-I promotes humoral (auto-)immunity: Autoantibodies, sometimes associated even with clinical signs of autoimmunity, occur in about 4–19% of patients with various diseases treated with IFN-α, and to lesser extent, also in multiple sclerosis during IFN-β therapy (Blake and Murphy, 1997; Durelli et al., 1999; Bitsch et al., 2004; Dionisiotis et al., 2004; Alanoglu et al., 2007; Borg and Isenberg, 2007). IFN-I-induced BAFF may synergize with other IFN-I-mediated effects established in animal models, including IFN-I-mediated stimulation of dendritic cells (Le Bon et al., 2001; Pascual et al., 2006), and direct effects of IFN-I on B cells (Le Bon and Pascual, 2006).
In SLE, a prototypical B cell-dependent systemic autoimmune disorder, IFN-I is elevated and considered to act as a major pathogenic factor (Cheema et al., 2001). Our finding that in multiple sclerosis, exogenously applied therapeutic IFN-I stimulates systemic BAFF levels in vivo might help to explain why in SLE, endogenously produced IFN-I stimulates systemic BAFF levels. Indeed, patients with SLE show elevated BAFF levels, which are likely to have pathogenic importance (MacKay et al., 1999).
Our ex vivo and in vitro studies identify monocytes, neutrophils, fibroblasts and astrocytes as strong BAFF producers in response to IFN-β, extending previous studies about BAFF induction by inflammatory cytokines in different cell types (Nardelli et al., 2001; Litinskiy et al., 2002; Krumbholz et al., 2005a; Ohata et al., 2005; Scapini et al., 2005; Ittah et al., 2006; Kato et al., 2006). Notably, BAFF was induced in these cells in vitro at IFN-β concentrations (about 1–10 U/ml) that were similar to the concentrations reached in vivo in treated multiple sclerosis patients (5–30 U/ml) (Mäurer et al., 2001). The observed induction of BAFF in astrocytes is especially interesting in view of our previous finding that astrocytes are a strong source of BAFF in lesions of untreated multiple sclerosis patients (Krumbholz et al., 2005a). It cannot be excluded (and might even be expected) that some therapeutically applied IFN-β might reach at least some CNS lesions where there is a locally disturbed blood–brain barrier. This would augment the pre-existing level of local BAFF production by astrocytes, turning the CNS into an even more ‘B cell-friendly milieu’. In those patients in whom B cells play a major important role, this would be a quite undesirable consequence of IFN-β therapy (see below).
Presently, it is impossible to predict, and difficult to assess whether an individual patient is likely to benefit from IFN-β therapy. The immunopathology of multiple sclerosis is likely heterogeneous. At least some patients are characterized by the production of pathogenic antibodies (Lucchinetti et al., 2000; Lucchinetti et al., 2002; Keegan et al., 2005; Breij et al., 2008). This has therapeutic implications: deposition of antibodies and complement in active lesions classified as pattern II lesions (Lucchinetti et al., 2000) have been linked to the therapeutic success of plasmapheresis (Keegan et al., 2005). A high B cell/monocyte ratio in the CSF seemed to predict a more progressive course (Cepok et al., 2001). When viewed in connection with our findings, there is concern that in patients with ‘B cell-dominated multiple sclerosis’ IFN-β treatment might further stimulate pathogenic B cell activity by upregulating systemic and potentially also local BAFF production, thus stimulating rather than inhibiting autoreactive B cells, autoantibody production and antigen presentation. One can speculate that such patients should benefit more from B cell targeting therapies such as anti-CD20 (rituximab) (Stuve et al., 2005; Antel and Bar-Or, 2006; Meinl et al., 2006; Hauser et al., 2008) and TACI-Ig (atacicept) (Hemmer and Hartung, 2007). Indeed, in neuromyelitis optica (NMO), an inflammatory CNS disease where there is concrete evidence for an antibody-mediated pathogenesis (Lucchinetti et al., 2002; Takahashi et al., 2007), IFN-β seems to be relatively ineffective or even harmful (Warabi et al., 2007). By contrast, rituximab therapy seemed to achieve a remarkable stabilization of NMO in a small study (Cree et al., 2005).
It should be kept in mind that B cells may also have beneficial effects on CNS autoimmunity, e.g. by secreting anti-inflammatory cytokines (Meinl et al., 2006). Whether and to what extent IFN-β promotes such beneficial effects, and whether and under what circumstances such beneficial effects can outweigh the presumed adverse effect of IFN-β-induced BAFF, is presently completely unknown. But this is clearly an important question that deserves to be studied in future investigations.
A well-known complication of IFN-β therapy in multiple sclerosis is the development of neutralizing antibodies which are directed against the injected IFN-β, and reduce its clinical efficacy (Kappos et al., 2005). One could speculate that development of neutralizing antibodies might be facilitated by BAFF induced by the therapeutically applied IFN-β. This question is difficult to study in a cross-sectional design, because in patients with high levels of neutralizing antibodies, the bioavailability of IFN-β would be decreased or lost. Trivially, therefore, IFN-induced BAFF levels would also be low. To properly study the possible effects of IFN-induced BAFF on the development of neutralizing antibodies would require a prospective design, demonstrating that high levels of BAFF precede the occurrence of neutralizing antibodies.
We found similar BAFF serum protein concentrations in untreated multiple sclerosis patients and healthy controls. It appears that BAFF systemic serum levels are mainly increased in systemic autoimmune diseases like SLE, but not necessarily in organ-specific diseases like multiple sclerosis, primary biliary cirrhosis and diabetes (Mackay et al., 2002). Our observation that the transcript levels of BAFF in whole blood and in separated monocytes were similar in untreated multiple sclerosis patients and controls contrasts somewhat with a previous publication which described elevated BAFF levels in blood monocytes of multiple sclerosis patients, but in that report the patients were not stratified for treatment (Thangarajh et al., 2004).
Our findings have implications for diseases other than multiple sclerosis. In the case of malignant melanoma, IFN-α therapy might exert beneficial effects also via BAFF induction and promotion of B cell function. Development of autoantibodies after IFN-α therapy in these patients is a positive prognostic marker for survival (Gogas et al., 2006). This indicates that in this case an increased immune function may be beneficial even at the expense of autoimmune side effects. IFN-α is also approved for therapy of hairy cell leukaemia and follicular lymphoma. Both are derived from the B cell linage and usually express BAFF-R (Nakamura et al., 2005; Rodig et al., 2005), which has been suggested to support the growth of these tumour cells (He et al., 2004; Nakamura et al., 2005; Rodig et al., 2005). The induction of BAFF by IFN-α therapy is therefore likely to counteract the beneficial effects of IFN-α by providing additional survival signals to the malignant B cells, thus limiting therapeutic success. Therefore, an additional BAFF blockade may increase the efficacy of IFN-α therapy in these B cell malignancies.
Concluding remarks
Our study reveals a complex situation: IFN-β therapy induces a potent B cell survival factor, BAFF, whereas B cell depletion is desirable at least in some multiple sclerosis patients. Of note, B cells themselves can exert both detrimental and beneficial effects. The systemic induction of BAFF by IFN-β therapy might facilitate the occurrence of various autoantibodies and IFN-neutralizing antibodies. Individual MS/NMO patients with evidence for a significant role of B cells do not appear to be ideal candidates for IFN-β therapy. This might partially explain inter-individual differences in the therapeutic response. It will be a major challenge for future investigations to define patient subgroups to predict who will or will not benefit from IFN-β therapy.
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
We are grateful to K. Korn and colleagues in the Institute of Virology, University of Erlangen, for providing HFF cells, and to Susan Kalled (Biogen-Idec) for providing BAFF ELISA reagents. We thank R. Rupec, M. Volkenandt, T. Maier and T. Vogt for patient samples. We thank Drs. A. Flügel and M. Kerschensteiner for their valuable comments on the manuscript. R.H. and E.M. have received grant support and/or consultancy fees from Schering, Teva, Serono, and Biogen-Idec. The other authors report no conflicts of interest. This work was funded by Deutsche Forschungsgemeinschaft (DFG SFB 571, in part grant TH 894); Verein zur Therapieforschung für MS-Kranke; Hermann and Lilly Schilling Foundation.
References
Abbreviations:
- APRIL
a proliferation-inducing ligand
- BAFF
B cell activating factor of the TNF family
- IFN
Interferon
- Ig
Immunoglobulin
- PBMC
peripheral blood mononuclear cell
- SLE
systemic lupus erythematosus.
Author notes
*These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}