




Abstract
As part of a biomarker discovery effort in peripheral blood, we acquired an immunological profile of cell-surface markers from healthy control and untreated subjects with relapsing–remitting MS (RRMS). Fresh blood from each subject was screened ex vivo using a panel of 50 fluorescently labelled monoclonal antibodies distributed amongst 56 pools of four antibodies each. From these 56 pools, we derived an immunological profile consisting of 1018 ‘features’ for each subject in our analysis using a systematic gating strategy. These profiles were interrogated in an analysis with a screening phase (23 patients) and an extension phase (15 patients) to identify cell populations in peripheral blood whose frequency is altered in untreated RRMS subjects. A population of CD8lowCD4− cells was identified as being reduced in frequency in untreated RRMS subjects (P = 0.0002), and this observation was confirmed in an independent sample of subjects from the Comprehensive Longitudinal Investigation of MS at the Brigham & Women's Hospital (P = 0.002). This reduction in the frequency of CD8lowCD4− cells is also observed in 38 untreated subjects with a clinically isolated demyelination syndrome (CIS) (P = 0.0006). We also show that these differences may be due to a reduction in the CD8lowCD56+CD3−CD4− subset of CD8low cells, which have a natural killer cell profile. Similarities between untreated CIS and RRMS subjects extend to broader immunological profiles: consensus clustering of our data suggests that there are three distinct populations of untreated RRMS subjects and that these distinct phenotypic categories are already present in our sample of untreated CIS subjects. Thus, our large-scale immunophenotyping approach has yielded robust evidence for a reduction of CD8lowCD4− cells in both CIS and RRMS in the absence of treatment as well as suggestive evidence for the existence of immunologically distinct subsets of subjects with a demyelinating disease.
Introduction
Profiling of peripheral blood has received much of the attention in biomarker discovery since this tissue is easily sampled and may reflect the manifestation of a disease process that occurs within tissues for which biopsy is not practical (e.g. CNS tissue). One illustration of this approach is the discovery of an interferon response signature in the peripheral blood mononuclear cells (PBMCs) of systemic lupus erythematosus patients (Baechler et al., 2003). However, the RNA expression profiling approach used in that study has not yet yielded a robust profile for multiple sclerosis (MS), an inflammatory demyelinating disease of the central nervous system (De Jager and Hafler, 2004). We have chosen to apply a flow cytometric analysis approach to biomarker discovery because it produces data at the single cell level and therefore enables us to assess differences in cell populations within a sample of blood and to measure the distribution of markers within each cell population.
Cytometric interrogation using combinations of antigens has been performed for decades in MS, starting with the first application of anti-CD4 and anti-CD8 antibodies that highlighted a reduced CD4/CD8 ratio in MS (Reinherz et al., 1980). However, there is, as yet, no diagnostic test that is clinically useful in MS, and the disease remains a diagnosis of exclusion. Application of cytometric analysis on a large scale is just now beginning but has already shown promise, as illustrated in a recent study that identified markers correlating with MS disease activity on magnetic resonance imaging (MRI) (Rinaldi et al., 2006). We have applied a large-scale cytometric approach to investigate peripheral blood from untreated subjects with relapsing–remitting MS (RRMS). Second, we have also analysed blood from subjects with a clinically isolated demyelinating syndrome (CIS). CIS is defined by the occurrence of a single episode of inflammatory demyelination (Miller et al., 2005), and many subjects with CIS go on to demonstrate clinical or imaging evidence of a second episode of demyelination and therefore fulfill a diagnosis of MS (McDonald et al., 2001). We report that both CIS and RRMS subjects demonstrate a reduction in the frequency of CD8lowCD4− cells when compared to healthy control subjects. This cell population is one that is increased in frequency with daclizumab treatment in MS (Bielekova et al., 2006). These cytometric profiles of peripheral blood have also uncovered the population structure of our patient sample, a structure consisting of three subgroups of subjects with RRMS or CIS. These observations further highlight the shared pathophysiology of these two clinical entities.
Material and Methods
Study design
This study uses data acquired in the MS Registry, a project that includes the prospective collection of flow cytometric data from untreated subjects with RRMS and healthy control subjects. Our study consists of a screening phase (Phase 1) in which cell populations are identified as demonstrating evidence of association to a diagnosis of MS. This screen was followed by an extension of the study (Phase 2), in which additional subjects were analysed to identify those cell populations presenting robust differences between healthy control subjects and subjects with MS. In Phase 3, a different flow cytometric data set, generated by the Comprehensive Longitudinal Investigation of MS at the Brigham & Women's Hospital (CLIMB), was used to independently replicate the major finding of the MS Registry Phase 2 analysis.
Secondary analyses were performed to explore the frequency of a cell population in subjects with CIS, to analyse the population structure of the patient samples, and to predict a diagnosis of MS.
Human subjects
In the screening phase (Phase 1 of the study), 23 untreated RRMS subjects and 17 healthy control subjects were assayed, and to validate results from our screen, we assayed an additional 15 untreated RRMS subjects and 15 healthy control subjects in the extension phase of the analysis (Phase 2). Another 11 subjects with CIS were recruited in parallel with the screening phase of the MS study (Fig. 1). All untreated subjects with RRMS met the following inclusion criteria: (i) age greater than 18 years; (ii) diagnosis of MS per the McDonald criteria (McDonald et al., 2001); (iii) no disease-modifying treatment or steroids in the preceding 4 weeks (steroids), 12 weeks (Glatiramer Acetate, Interferon β1a or β1b and methotrexate) or 24 weeks (cytoxan and mitoxantrone); and (iv) no evidence of progressive disease, as defined by progressive functional decline with deficits lasting 6 months or more. There were no exclusion criteria for subjects meeting the inclusion criteria. The untreated subjects with CIS met the same inclusion criteria, except for (ii): instead, these subjects with CIS had a history of a single episode of inflammatory demyelination documented by a neurologist and had two or more periventricular or ovoid hyperintense T2 lesions of >3 mm on MRI (Miller et al., 2005). All subjects were recruited sequentially through the Partners MSCenter in Boston, MA. All RRMS subjects and 11 CIS subjects have a complete immunophenotypic profile; data on these subjects were collected in a single time window that overlaps data collection for control subjects. An additional 27 CIS subjects were recruited later using the same inclusion criteria but only data from pool ‘a1’ (Supplementary Table 1a) was gathered from these individuals. All of the untreated subjects with MS or CIS that were enrolled into the MS Registry were used in the analysis; no subject was excluded. The untreated subjects with RRMS include subjects who had either declined treatment or were in between treatment for their MS. All CIS and RRMS subjects were recruited into the MS Registry at the time of a routine clinical visit scheduled every 6 months to monitor changes in the clinical examination of our patients. Healthy control subjects were recruited through the Partners Healthcare RSVP for Health database (http://www.rsvpforhealth.org/) of individuals who are interested in participating in medical research. All healthy control subjects are over 18 years of age. All subjects are of self-declared European (‘white’) ancestry. The clinical features of our subject sample are presented in Table 1.
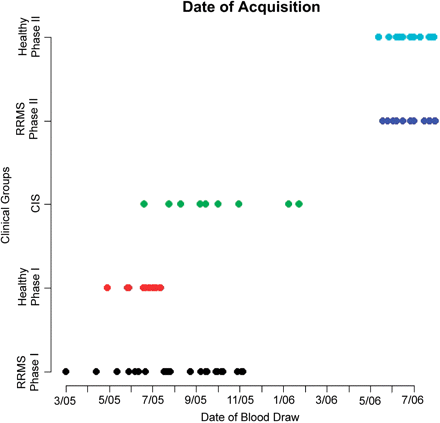
Distribution of the dates of blood draws for all subjects with a complete immunological profile that are included in our analysis. Phase 1 subjects refer to those subjects used in screening phase of our analysis; Phase 2 subjects are the additional subjects collected for the extension analysis Colours: Black (RRMS Phase 1), Red ((Healthy Control Phase 1), Green (CIS), Navy Blue (RRMS Phase 2), and Light Blue (Healthy Control Phase 2).
Clinical features of subjects included in our analyses.
| Clinical variable | MS Registry | CLIMB | |||||
|---|---|---|---|---|---|---|---|
| Phase 1 MS n = 23 | Phase 1 Healthy n = 17 | Phase 2 MS n = 15 | Phase 2 Healthy n = 15 | CIS n = 38 | Phase 3 MS n = 16 | Phase 3 Healthy n = 18 | |
| Mean age | 40.4 | 46.8 | 43.9 | 39.2 | 35.5 | 44.9 | 37.3 |
| Gender ratio (F:M) | 1.88:1 | 7.5:1 | 1.5:1 | 1.5:1 | 7:1 | 7:1 | 8:1 |
| Disease duration | 10.4 | – | 12.9 | – | 2.7 | 8.6 | – |
| Mean age of symptom onset | 30.0 | – | 31.0 | – | 32.7 | 36.3 | – |
| Mean EDSS score | 1.43 | – | 1.53 | – | 0.77 | 1.07 | – |
| Benign MS (%) | 34.7 | – | 13.3 | – | – | 12.5 | – |
| Clinical variable | MS Registry | CLIMB | |||||
|---|---|---|---|---|---|---|---|
| Phase 1 MS n = 23 | Phase 1 Healthy n = 17 | Phase 2 MS n = 15 | Phase 2 Healthy n = 15 | CIS n = 38 | Phase 3 MS n = 16 | Phase 3 Healthy n = 18 | |
| Mean age | 40.4 | 46.8 | 43.9 | 39.2 | 35.5 | 44.9 | 37.3 |
| Gender ratio (F:M) | 1.88:1 | 7.5:1 | 1.5:1 | 1.5:1 | 7:1 | 7:1 | 8:1 |
| Disease duration | 10.4 | – | 12.9 | – | 2.7 | 8.6 | – |
| Mean age of symptom onset | 30.0 | – | 31.0 | – | 32.7 | 36.3 | – |
| Mean EDSS score | 1.43 | – | 1.53 | – | 0.77 | 1.07 | – |
| Benign MS (%) | 34.7 | – | 13.3 | – | – | 12.5 | – |
Phase 1 subjects were used in our initial screen of flow cytometric features in the MS Registry. Phase 2 subjects include those subjects recruited at a later time and added to the Phase 1 subjects for the extension analysis. Eleven CIS subjects were collected in parallel with Phase 1 subjects and have a complete immunologic profile of 1018 features; an additional 27 CIS subjects were collected later and only the CD8low cell population was measured in these subjects. There is no significant difference between the demographics of the two groups of CIS subjects. Phase 3 subjects were recruited as part of a different study, CLIMB. Benign MS is defined as an EDSS score <2 at 10 years after the onset of symptoms. Glossary: F = female; M = male.
Clinical features of subjects included in our analyses.
| Clinical variable | MS Registry | CLIMB | |||||
|---|---|---|---|---|---|---|---|
| Phase 1 MS n = 23 | Phase 1 Healthy n = 17 | Phase 2 MS n = 15 | Phase 2 Healthy n = 15 | CIS n = 38 | Phase 3 MS n = 16 | Phase 3 Healthy n = 18 | |
| Mean age | 40.4 | 46.8 | 43.9 | 39.2 | 35.5 | 44.9 | 37.3 |
| Gender ratio (F:M) | 1.88:1 | 7.5:1 | 1.5:1 | 1.5:1 | 7:1 | 7:1 | 8:1 |
| Disease duration | 10.4 | – | 12.9 | – | 2.7 | 8.6 | – |
| Mean age of symptom onset | 30.0 | – | 31.0 | – | 32.7 | 36.3 | – |
| Mean EDSS score | 1.43 | – | 1.53 | – | 0.77 | 1.07 | – |
| Benign MS (%) | 34.7 | – | 13.3 | – | – | 12.5 | – |
| Clinical variable | MS Registry | CLIMB | |||||
|---|---|---|---|---|---|---|---|
| Phase 1 MS n = 23 | Phase 1 Healthy n = 17 | Phase 2 MS n = 15 | Phase 2 Healthy n = 15 | CIS n = 38 | Phase 3 MS n = 16 | Phase 3 Healthy n = 18 | |
| Mean age | 40.4 | 46.8 | 43.9 | 39.2 | 35.5 | 44.9 | 37.3 |
| Gender ratio (F:M) | 1.88:1 | 7.5:1 | 1.5:1 | 1.5:1 | 7:1 | 7:1 | 8:1 |
| Disease duration | 10.4 | – | 12.9 | – | 2.7 | 8.6 | – |
| Mean age of symptom onset | 30.0 | – | 31.0 | – | 32.7 | 36.3 | – |
| Mean EDSS score | 1.43 | – | 1.53 | – | 0.77 | 1.07 | – |
| Benign MS (%) | 34.7 | – | 13.3 | – | – | 12.5 | – |
Phase 1 subjects were used in our initial screen of flow cytometric features in the MS Registry. Phase 2 subjects include those subjects recruited at a later time and added to the Phase 1 subjects for the extension analysis. Eleven CIS subjects were collected in parallel with Phase 1 subjects and have a complete immunologic profile of 1018 features; an additional 27 CIS subjects were collected later and only the CD8low cell population was measured in these subjects. There is no significant difference between the demographics of the two groups of CIS subjects. Phase 3 subjects were recruited as part of a different study, CLIMB. Benign MS is defined as an EDSS score <2 at 10 years after the onset of symptoms. Glossary: F = female; M = male.
In Phase 2 of the study, a more detailed matching protocol was instituted: each healthy control subject was matched on gender and age within 5 years to a subject with MS. In addition, healthy control blood samples were collected within two days of the sampling of the matched subject with MS. All of the healthy control subjects used in Phase 2 were also recruited from the Partners Healthcare RSVP for Health resource and were of self-declared European ancestry. The inclusion and exclusion criteria remained the same (see the Phase 1 criteria described earlier).
The independent replication sample set used in Phase 3 was drawn from data gathered prospectively as part of the CLIMB. These subjects with MS were also in the relapsing–remitting phase of their disease and were untreated at the time of sampling. All of these subjects met the McDonald criteria for a diagnosis of MS (McDonald et al., 2001) as well as the inclusion criteria described for the Phase 1 described earlier. The clinical profile of this subject sample is presented in Table 1. The healthy control subjects in this case were collected from laboratory personnel who are self-reported as being free of inflammatory disease. All of the CLIMB subjects used here are of self-declared European ancestry.
All MS subjects analysed in this study (whether they were enrolled in the MS Registry or CLIMB) were recruited from the Partners MS Center in Boston. None of the CLIMB subjects are duplicates of the MS Registry subjects.
Human samples
At the time of enrolment into the MS Registry, ∼7 ml of blood was collected from each subject and processed to capture flow cytometric data. Samples were collected between 09:00 and 15:00 from our Partners MS Center in Boston, MA, USA. In addition, 15 of the 17 healthy control subjects enrolled in Phase 1 were assayed serially: at enrollment, 6 months later, and finally 12 months later. This longitudinal component of the project was used in our quality control (QC) analysis to select the flow cytometric parameters that are robust to variation over time and would therefore be more informative in an analysis for which the samples were collected over 18 months. Each subject's blood sample underwent staining with the fluorescently labelled antibody pools within 2 h of blood collection. Sample staining, processing and fixation were completed within 4 h of blood collection; the median time to flow cytometry was 6 h following sample collection, with a range of 4–24 h. The details of the staining and flow cytometry procedures are presented below.
CLIMB blood samples were likewise completely processed, stained and fixed within 4 h of phlebotomy. Flow cytometry was performed within 24 h of sample collection.
MS Registry sample preparation and flow cytometry
Samples of whole blood were stained with each combination of four fluorescently conjugated monoclonal antibodies listed in Supplementary Table 1a. The source of each antibody is listed in Supplementary Table 1b. The four antibodies in the pools of each well were pre-mixed and diluted to a final volume of 1.5 ml, and batches of approximately 100 plates were prepared by distributing 15 μl of each antibody pool to the corresponding wells. The amount of each of the antibodies in each well was typically either 1.5 or 2.5 μl. Antibody pool plates were typically used within 2 months and kept tightly sealed at 4°C until used. 85 μl of blood were added to wells, mixed and incubated at ambient temperature for 10 min. After incubation, cells were washed twice with Dulbecco's phosphate-buffered saline (w/o Ca2+ and Mg2+), and red blood cells were lysed. Cells were then fixed by distributing 180 μl of BD fluorescence activated cell sorter (FACS) Lysis Buffer to the wells and incubating at 37°C for 10 min prior to centrifugation. Cells were then re-suspended in 100 μl of BD FACS Lysis Buffer and transferred to Costar cluster tubes for data acquisition by flow cytometry. Samples were analysed by flow cytometry using a FACSCalibur™ FACS and CellQuest™ Pro software (both from BD Biosciences, San Jose, CA, USA). Total event collection ceased when 30 000 events were collected in the total leukocyte gate.
CLIMB sample preparation and flow cytometry
Blood samples were collected at the Partners MS Center, and processing was completed within 4 h. PBMCs were isolated by centrifugation over Ficoll–Hypaque density gradient and washed 3 times in PBS containing 1% fetal calf serum. After the final wash, cells were re-suspended in Staining Buffer (100 000 cells/100 μl). Cells were incubated for 20 min with different combinations of fluorescent antibodies then washed twice and re-suspended in staining buffer for data acquisition by FACSCalibur (Beckman Dickinson, BD). CD4 FITC-labelled and CD8 Cychrome-labelled antibodies were purchased from BD Biosciences (cat#555246 and 555368, respectively).
Data pre-processing
We used the FlowJo software suite v6.4.2 (Treestar, Ashland OR, USA) to extract 5 parameters from the MS registry and CLIMB data: for each gated cell population, we captured (i) the frequency of the gated cells in the parental cell population, (ii) mean fluorescence intensity (MFI), (iii) median fluorescence intensity (MedFI), (iv) mean channel value (MCV) and (v) geometric mean fluorescence intensity (GMFI).
The systematic gating protocol we followed is as follows: data from each blood sample are first projected in two dimensions (forward and side scatter), and three gates were placed as illustrated by Supplementary Fig. 1. Gate 1 captures mostly lymphocyte populations; Gate 2 captures monocytes and activated lymphocytes; and Gate 3 captures predominantly granulocyte populations. Each gated cell population is then projected onto two dimensions using two of the four antibodies found in each pool. Using isotype control data, quadrants are placed to define positive and negative cell populations for each dimension, and data are acquired from each quadrant. For example, the proportion of Gate 1 cells that are CD4 positive and CXCR2 negative was captured as part of the data acquisition on pool ‘b1’ (Supplementary Table 1a); in our database, this datum is stored with the label ‘Gate 1/CD4posCXCR2neg’ to describe its source. Some antibody combinations are found in multiple pools, so that a number is attached to the end of the label to differentiate the antibody pool from which it was extracted (e.g. ‘Gate 1/CD4posCCR7pos4’, the fourth independent assessment of this antibody combination).
Cell populations found in each of the four quadrants from a 2D projection were then projected in the remaining two dimensions (two antibodies) of each pool, and the process of placing quadrants based on isotype data was repeated. Data were then extracted from each quadrant to produce data features such as ‘Gate 1/CD19posCD4neg/CD14negCCR4pos’.
Thus, we instituted a systematic data extraction protocol from each quadrant of successive 2D projections of our data. Each antibody pool had data collected using a single sequence of 2D projections. Many quadrants therefore had few or no cells, and we address the use of such data in our QC analysis (see subsequently). Specifically, quadrants with 30 or fewer cells were excluded from the analysis pipeline since the quantity of cells was deemed too small to generate robust data. A final check was implemented on the results of the extension analysis: if a feature originates from a gate that does not capture a clear, unique cell population, it was not included in the final results table, Table 2. One of the two features that failed this criterion is illustrated in Supplementary Table 2b.
Cell populations whose frequency is different in our comparison of untreated RRMS to healthy control subjects.
| Cell subset | Exploration P-value | Extension P-value | Exploration Healthy mean (%) | Exploration MS mean (%) | Extension Healthy mean (%) | Extension MS mean (%) |
|---|---|---|---|---|---|---|
| CD8low cell population | ||||||
| Gate 1/CD8lowCCR1neg | 0.0539 | 0.0008 | 6.99 | 5.57 | 7.54 | 5.57 |
| Gate 1/CD8lowCCR2neg | 0.0199 | 0.0016 | 6.98 | 5.09 | 7 | 5.01 |
| Gate 1/CD8lowCCR3neg | 0.0184 | 0.0005 | 7.07 | 5.31 | 7.35 | 5.38 |
| Gate 1/CD8lowCCR5neg | 0.0269 | 0.0006 | 6.55 | 5.15 | 6.95 | 5.08 |
| Gate 1/CD8lowCCR5neg2 | 0.0139 | 0.0006 | 6.54 | 5.11 | 6.66 | 4.9 |
| Gate 1/CD8lowCCR7neg | 0.0109 | 0.0013 | 6.49 | 4.77 | 6.32 | 4.45 |
| Gate 1/CD8lowCD14neg | 0.0555 | 0.0013 | 7.44 | 5.97 | 8.07 | 6.13 |
| Gate 1/CD8lowCD14neg/CD4negCCR5neg | 0.0835 | 0.0162 | 87.89 | 81.08 | 87.09 | 76.78 |
| Gate 1/CD8lowCD14neg2 | 0.0163 | 0.0009 | 7.49 | 5.52 | 7.91 | 5.84 |
| Gate 1/CD8lowCD14neg3 | 0.0674 | 0.0027 | 7.53 | 5.8 | 8.06 | 6.06 |
| Gate 1/CD8lowCD27neg | 0.0295 | 0.0006 | 4.84 | 3.67 | 5.38 | 3.84 |
| Gate 1/CD8lowCD27neg2 | 0.0697 | 0.002 | 4.71 | 3.68 | 5.26 | 3.86 |
| Gate 1/CD8lowCD27neg3 | 0.074 | 0.0025 | 4.83 | 3.72 | 5.16 | 3.71 |
| Gate 1/CD8lowCD27neg4 | 0.0623 | 0.0034 | 4.86 | 3.78 | 5.44 | 3.96 |
| Gate 1/CD8lowCD3neg | 0.0215 | 0.0041 | 5.49 | 3.97 | 6.59 | 4.73 |
| Gate 1/CD8lowCD4neg | 0.0188 | 0.0011 | 7.08 | 5.36 | 8.11 | 5.71 |
| Gate 1/CD8lowCD4neg2 | 0.0707 | 0.0007 | 6.73 | 5.47 | 7.55 | 5.42 |
| Gate 1/CD8lowCD4neg3* | 0.0126 | 0.0002 | 6.76 | 5 | 7.31 | 5.12 |
| Gate 1/CD8lowCD4neg4 | 0.0427 | 0.0005 | 6.89 | 5.29 | 7.51 | 5.31 |
| Gate 1/CD8lowCD56pos | 0.0347 | 0.0052 | 6.4 | 4.89 | 7.19 | 5.46 |
| CD4 cell population | ||||||
| Gate 1/CD4posCD19neg4 | 0.0419 | 0.0164 | 45.56 | 50.91 | 42.86 | 48.35 |
| Gate 1/CD4posCXCR2neg | 0.034 | 0.0043 | 43.16 | 50.04 | 40.46 | 47.77 |
| Cell subset | Exploration P-value | Extension P-value | Exploration Healthy mean (%) | Exploration MS mean (%) | Extension Healthy mean (%) | Extension MS mean (%) |
|---|---|---|---|---|---|---|
| CD8low cell population | ||||||
| Gate 1/CD8lowCCR1neg | 0.0539 | 0.0008 | 6.99 | 5.57 | 7.54 | 5.57 |
| Gate 1/CD8lowCCR2neg | 0.0199 | 0.0016 | 6.98 | 5.09 | 7 | 5.01 |
| Gate 1/CD8lowCCR3neg | 0.0184 | 0.0005 | 7.07 | 5.31 | 7.35 | 5.38 |
| Gate 1/CD8lowCCR5neg | 0.0269 | 0.0006 | 6.55 | 5.15 | 6.95 | 5.08 |
| Gate 1/CD8lowCCR5neg2 | 0.0139 | 0.0006 | 6.54 | 5.11 | 6.66 | 4.9 |
| Gate 1/CD8lowCCR7neg | 0.0109 | 0.0013 | 6.49 | 4.77 | 6.32 | 4.45 |
| Gate 1/CD8lowCD14neg | 0.0555 | 0.0013 | 7.44 | 5.97 | 8.07 | 6.13 |
| Gate 1/CD8lowCD14neg/CD4negCCR5neg | 0.0835 | 0.0162 | 87.89 | 81.08 | 87.09 | 76.78 |
| Gate 1/CD8lowCD14neg2 | 0.0163 | 0.0009 | 7.49 | 5.52 | 7.91 | 5.84 |
| Gate 1/CD8lowCD14neg3 | 0.0674 | 0.0027 | 7.53 | 5.8 | 8.06 | 6.06 |
| Gate 1/CD8lowCD27neg | 0.0295 | 0.0006 | 4.84 | 3.67 | 5.38 | 3.84 |
| Gate 1/CD8lowCD27neg2 | 0.0697 | 0.002 | 4.71 | 3.68 | 5.26 | 3.86 |
| Gate 1/CD8lowCD27neg3 | 0.074 | 0.0025 | 4.83 | 3.72 | 5.16 | 3.71 |
| Gate 1/CD8lowCD27neg4 | 0.0623 | 0.0034 | 4.86 | 3.78 | 5.44 | 3.96 |
| Gate 1/CD8lowCD3neg | 0.0215 | 0.0041 | 5.49 | 3.97 | 6.59 | 4.73 |
| Gate 1/CD8lowCD4neg | 0.0188 | 0.0011 | 7.08 | 5.36 | 8.11 | 5.71 |
| Gate 1/CD8lowCD4neg2 | 0.0707 | 0.0007 | 6.73 | 5.47 | 7.55 | 5.42 |
| Gate 1/CD8lowCD4neg3* | 0.0126 | 0.0002 | 6.76 | 5 | 7.31 | 5.12 |
| Gate 1/CD8lowCD4neg4 | 0.0427 | 0.0005 | 6.89 | 5.29 | 7.51 | 5.31 |
| Gate 1/CD8lowCD56pos | 0.0347 | 0.0052 | 6.4 | 4.89 | 7.19 | 5.46 |
| CD4 cell population | ||||||
| Gate 1/CD4posCD19neg4 | 0.0419 | 0.0164 | 45.56 | 50.91 | 42.86 | 48.35 |
| Gate 1/CD4posCXCR2neg | 0.034 | 0.0043 | 43.16 | 50.04 | 40.46 | 47.77 |
The table lists those features for which evidence of association with RRMS was enhanced after the inclusion of additional samples in the extension analysis and whose extension P-value was <0.05. The features are arranged by alphabetical order within the two cell populations (CD8low and CD4+) that we identify as being found at different frequency in cases and controls within our screen. We also present the mean frequency value of each selected feature at both stages of the analysis in the healthy control and RRMS subjects. The ‘exploration’ analysis is performed with only Phase 1 subjects; the ‘extension’ analysis is performed in the combined samples of Phase 1 and 2. The ‘neg’ is used to denote cell populations for which a particular marker is absent; ‘pos’ is used to denote cell populations positive for a particular marker. *Some features were independently measured in different wells of the experimental plates. When a feature has a number as its last character, the number refers to the particular iteration of that feature on the experimental plate. For example, ‘CD8lowCD4neg3’ is the third of four independent measurements of the CD8lowCD4neg population.
Cell populations whose frequency is different in our comparison of untreated RRMS to healthy control subjects.
| Cell subset | Exploration P-value | Extension P-value | Exploration Healthy mean (%) | Exploration MS mean (%) | Extension Healthy mean (%) | Extension MS mean (%) |
|---|---|---|---|---|---|---|
| CD8low cell population | ||||||
| Gate 1/CD8lowCCR1neg | 0.0539 | 0.0008 | 6.99 | 5.57 | 7.54 | 5.57 |
| Gate 1/CD8lowCCR2neg | 0.0199 | 0.0016 | 6.98 | 5.09 | 7 | 5.01 |
| Gate 1/CD8lowCCR3neg | 0.0184 | 0.0005 | 7.07 | 5.31 | 7.35 | 5.38 |
| Gate 1/CD8lowCCR5neg | 0.0269 | 0.0006 | 6.55 | 5.15 | 6.95 | 5.08 |
| Gate 1/CD8lowCCR5neg2 | 0.0139 | 0.0006 | 6.54 | 5.11 | 6.66 | 4.9 |
| Gate 1/CD8lowCCR7neg | 0.0109 | 0.0013 | 6.49 | 4.77 | 6.32 | 4.45 |
| Gate 1/CD8lowCD14neg | 0.0555 | 0.0013 | 7.44 | 5.97 | 8.07 | 6.13 |
| Gate 1/CD8lowCD14neg/CD4negCCR5neg | 0.0835 | 0.0162 | 87.89 | 81.08 | 87.09 | 76.78 |
| Gate 1/CD8lowCD14neg2 | 0.0163 | 0.0009 | 7.49 | 5.52 | 7.91 | 5.84 |
| Gate 1/CD8lowCD14neg3 | 0.0674 | 0.0027 | 7.53 | 5.8 | 8.06 | 6.06 |
| Gate 1/CD8lowCD27neg | 0.0295 | 0.0006 | 4.84 | 3.67 | 5.38 | 3.84 |
| Gate 1/CD8lowCD27neg2 | 0.0697 | 0.002 | 4.71 | 3.68 | 5.26 | 3.86 |
| Gate 1/CD8lowCD27neg3 | 0.074 | 0.0025 | 4.83 | 3.72 | 5.16 | 3.71 |
| Gate 1/CD8lowCD27neg4 | 0.0623 | 0.0034 | 4.86 | 3.78 | 5.44 | 3.96 |
| Gate 1/CD8lowCD3neg | 0.0215 | 0.0041 | 5.49 | 3.97 | 6.59 | 4.73 |
| Gate 1/CD8lowCD4neg | 0.0188 | 0.0011 | 7.08 | 5.36 | 8.11 | 5.71 |
| Gate 1/CD8lowCD4neg2 | 0.0707 | 0.0007 | 6.73 | 5.47 | 7.55 | 5.42 |
| Gate 1/CD8lowCD4neg3* | 0.0126 | 0.0002 | 6.76 | 5 | 7.31 | 5.12 |
| Gate 1/CD8lowCD4neg4 | 0.0427 | 0.0005 | 6.89 | 5.29 | 7.51 | 5.31 |
| Gate 1/CD8lowCD56pos | 0.0347 | 0.0052 | 6.4 | 4.89 | 7.19 | 5.46 |
| CD4 cell population | ||||||
| Gate 1/CD4posCD19neg4 | 0.0419 | 0.0164 | 45.56 | 50.91 | 42.86 | 48.35 |
| Gate 1/CD4posCXCR2neg | 0.034 | 0.0043 | 43.16 | 50.04 | 40.46 | 47.77 |
| Cell subset | Exploration P-value | Extension P-value | Exploration Healthy mean (%) | Exploration MS mean (%) | Extension Healthy mean (%) | Extension MS mean (%) |
|---|---|---|---|---|---|---|
| CD8low cell population | ||||||
| Gate 1/CD8lowCCR1neg | 0.0539 | 0.0008 | 6.99 | 5.57 | 7.54 | 5.57 |
| Gate 1/CD8lowCCR2neg | 0.0199 | 0.0016 | 6.98 | 5.09 | 7 | 5.01 |
| Gate 1/CD8lowCCR3neg | 0.0184 | 0.0005 | 7.07 | 5.31 | 7.35 | 5.38 |
| Gate 1/CD8lowCCR5neg | 0.0269 | 0.0006 | 6.55 | 5.15 | 6.95 | 5.08 |
| Gate 1/CD8lowCCR5neg2 | 0.0139 | 0.0006 | 6.54 | 5.11 | 6.66 | 4.9 |
| Gate 1/CD8lowCCR7neg | 0.0109 | 0.0013 | 6.49 | 4.77 | 6.32 | 4.45 |
| Gate 1/CD8lowCD14neg | 0.0555 | 0.0013 | 7.44 | 5.97 | 8.07 | 6.13 |
| Gate 1/CD8lowCD14neg/CD4negCCR5neg | 0.0835 | 0.0162 | 87.89 | 81.08 | 87.09 | 76.78 |
| Gate 1/CD8lowCD14neg2 | 0.0163 | 0.0009 | 7.49 | 5.52 | 7.91 | 5.84 |
| Gate 1/CD8lowCD14neg3 | 0.0674 | 0.0027 | 7.53 | 5.8 | 8.06 | 6.06 |
| Gate 1/CD8lowCD27neg | 0.0295 | 0.0006 | 4.84 | 3.67 | 5.38 | 3.84 |
| Gate 1/CD8lowCD27neg2 | 0.0697 | 0.002 | 4.71 | 3.68 | 5.26 | 3.86 |
| Gate 1/CD8lowCD27neg3 | 0.074 | 0.0025 | 4.83 | 3.72 | 5.16 | 3.71 |
| Gate 1/CD8lowCD27neg4 | 0.0623 | 0.0034 | 4.86 | 3.78 | 5.44 | 3.96 |
| Gate 1/CD8lowCD3neg | 0.0215 | 0.0041 | 5.49 | 3.97 | 6.59 | 4.73 |
| Gate 1/CD8lowCD4neg | 0.0188 | 0.0011 | 7.08 | 5.36 | 8.11 | 5.71 |
| Gate 1/CD8lowCD4neg2 | 0.0707 | 0.0007 | 6.73 | 5.47 | 7.55 | 5.42 |
| Gate 1/CD8lowCD4neg3* | 0.0126 | 0.0002 | 6.76 | 5 | 7.31 | 5.12 |
| Gate 1/CD8lowCD4neg4 | 0.0427 | 0.0005 | 6.89 | 5.29 | 7.51 | 5.31 |
| Gate 1/CD8lowCD56pos | 0.0347 | 0.0052 | 6.4 | 4.89 | 7.19 | 5.46 |
| CD4 cell population | ||||||
| Gate 1/CD4posCD19neg4 | 0.0419 | 0.0164 | 45.56 | 50.91 | 42.86 | 48.35 |
| Gate 1/CD4posCXCR2neg | 0.034 | 0.0043 | 43.16 | 50.04 | 40.46 | 47.77 |
The table lists those features for which evidence of association with RRMS was enhanced after the inclusion of additional samples in the extension analysis and whose extension P-value was <0.05. The features are arranged by alphabetical order within the two cell populations (CD8low and CD4+) that we identify as being found at different frequency in cases and controls within our screen. We also present the mean frequency value of each selected feature at both stages of the analysis in the healthy control and RRMS subjects. The ‘exploration’ analysis is performed with only Phase 1 subjects; the ‘extension’ analysis is performed in the combined samples of Phase 1 and 2. The ‘neg’ is used to denote cell populations for which a particular marker is absent; ‘pos’ is used to denote cell populations positive for a particular marker. *Some features were independently measured in different wells of the experimental plates. When a feature has a number as its last character, the number refers to the particular iteration of that feature on the experimental plate. For example, ‘CD8lowCD4neg3’ is the third of four independent measurements of the CD8lowCD4neg population.
Of note, the anti-CD8 antibody data was processed slightly differently. Prior to data extraction, we had decided to split the CD8 dimension into three components to be able to analyse data from the population of cells that express low levels of CD8 (CD8low) separately; an example of this gating strategy is shown in Fig. 2A. Finally, the measurements involving the anti-CXCR3 antibody had to be eliminated because the anti-CXCR3 antibody failed to stain one-half of the samples; this was due to a batch of bad antibody.
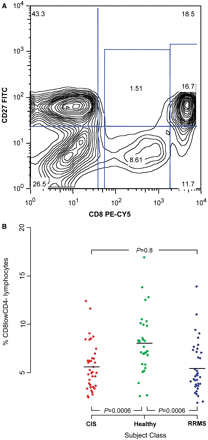
The CD8low population is decreased in frequency in untreated subjects with RRMS. (A) Representative density plot of a subject with RRMS outlining the gates used to capture the CD8low and CD8+ cell populations. These flow cytometric data were selected from ‘Gate 1’, the gate that captures the lymphocyte population in our sample (see supplementary Fig. 1). (B) We have plotted the frequency (%) of the CD8lowCD4− feature, the most differentiated feature in our analysis, for each of the healthy control (green), CIS (red) and RRMS (green) subjects. The frequency is calculated based on the number of cells found within Gate 1; thus, the denominator is the total number of cells found within our lymphocyte gate.
QC analysis
Fifteen of the healthy control subjects were assayed longitudinally, and these data were used to select the cytometric parameters that are robust to variation over time. Specifically, these 15 healthy control subjects were sampled at the time of enrolment, at 6 months, and at 12 months. At each time point, the peripheral blood was interrogated with the same panel of antibodies, and 5 parameters were captured for each gated cell population: frequency of the gated cells in the cell population found in the preceding gate in the sequence of data processing, MFI, MedFI, MCV and GMFI. Our systematic gating procedure included 4747 gates from which the five parameters were extracted.
To select the most robust parameters for our analysis, we followed the following QC pipeline. First, we eliminated data from all gates where the gated cell population consisted of less than 30 cells, as such low numbers of cells are unlikely to provide accurate estimates of marker expression in true subpopulations of PBMCs. This first processing step reduced the number of gates to 1018. Using these 1018 gated cell populations that met our initial QC criterion of cell count, we compared the measurements performed across the three time points (TP1-3) of each healthy control subject. We calculated a correlation coefficient (r) corresponding to the comparison of a given sample's feature vector to the two repeated feature vectors. Because of the lower mean correlation coefficients (r = 0.51–0.60) in the 4 out of the 5 parameters relating to fluorescence intensity, we have limited our analysis to the frequency variable which had a correlation coefficient across the three time points averaging r = 0.77 (range r = 0.52–0.91). This variability is likely to have both a technical component and a biological component. To minimize technical variability in our frequency parameter that may arise from differences amongst antibody batches and operators, we have included in our analysis only those MS and healthy subjects whose sample collection time overlapped during the course of the MS Registry project. Thus, in the screening phase of our analysis (Phase 1), we use the third time point (TP3) value for each healthy control in our comparison with subjects with MS since these control samples were collected during the time that the majority of the MS and CIS samples were collected (Fig. 1). The extension phase subjects were recruited using a strict clinical and temporal matching protocol that is described earlier. At the end of our QC process, we therefore had a reduced data set of 1018 different frequency measurements or ‘features’ for each of our 81 subjects; these features were then loaded into our analysis pipeline. Some of these features are captured in more than one well; for example, four wells have both CD4 and CD8 antibodies. When redundant features are removed, we are left with 795 features in our data set.
Duplicate wells were not part of the experimental design. However, we offer an estimate of the intra-plate variability of our measurement by analysing data from the four wells that have both the anti-CD4 and anti-CD8 antibodies. Details are presented in Supplementaryl Table 2, the mean correlation coefficient (r) is 0.87 (range 0.78–0.97).
Analysis pipeline
In collecting our results, we used an analysis pipeline that consists of three main steps. These steps are significance testing, dimensionality reduction and prediction model building. The three steps are derived from the GenePattern algorithms comparative marker selection (CMS), non-negative matrix factorization (NMF) and support vector machine (SVM) that have been tailored to work together in a cross-validation loop (http://www.broad.mit.edu/cancer/software/genepattern/) (Reich et al., 2006). The pipeline can be replicated using the GenePattern environment.
Significance testing for each frequency feature
In the primary analysis of our data, we use CMS to perform a permutation test on each feature to assess its correlation with class (untreated RRMS or healthy) and to quantify that correlation. The resulting P-value is reported for each feature in our data set. For the replication data set from CLIMB, we used a Wilcoxon exact test.
Prediction analyses
For the prediction analyses, CMS is included in a loop as follows: a random sample is removed from the data set, and the remaining data is introduced into the pipeline as an n × m matrix, where there are n features and m samples (m = M−1). Also associated with this matrix is a class file, indicating which of the total c classes to which each sample belongs. The matrix and class files are introduced into CMS which performs a permutation test on each feature to assess its correlation with class (untreated RRMS or healthy) and to quantify that correlation. The original n × m matrix is then reduced to a (cp) × m matrix, where p (specified by the user) represents the p most distinguishing features according to the permutation tests for each of the c classes.
The (cp) × m matrix is then introduced to the second component of the pipeline: NMF. The details of NMF are described in the NMF documentation on the GenePattern website (http://www.broad.mit.edu/cancer/software/genepattern/) (Reich et al., 2006). NMF finds patterns of feature expression in the data set. The final output is a k × m matrix, where k is the number of distinct patterns (‘meta-features’) set by the user and defined by the algorithm. We found the optimal value of k to be 3, as demonstrated using NMF consensus clustering (Fig. 3). The NMF consensus clustering is an additional algorithm published in Genepattern.
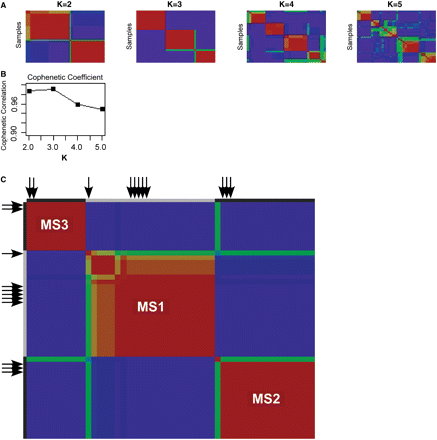
There are three subsets of subjects in our sample of untreated individuals with RRMS. (A) Consensus clustering of MS subjects into different numbers of clusters. All 38 of the RRMS samples are used in this analysis. ‘k’ denotes the number of clusters being tested; k = 2–5 were tested. The cophenetic coefficient is a measure of how well the number of clusters fits the data, with 1.0 being maximal. Each rectangle is a table, with each subject being listed in the same order on the X- and Y- axes, starting from the top left corner. Each cell of the table contains the probability that a pair of subjects is part of the same cluster. The likelihood that two subjects are part of the same cluster can be high (red colour) or moderate (yellow); a blue colour denotes the fact that a pair of subjects is unlikely to be part of the same cluster. A green colour denotes that the algorithm is uncertain and chooses not to make a call (B) We have plotted the cophenetic coefficient for each number or ‘k’ attempted and see that our data best fits a model with three separate subsets of subjects (k = 3). (C) We repeat the clustering analysis after adding 11 subjects with CIS to the pooled RRMS samples. The subjects with CIS are marked with a black arrow.
The last step in our pipeline is testing the k × m matrix for its ability to predict a random sample. The random sample is the original sample that we pulled out. We used a tailored version of a SVM to execute this prediction. We employed the SVM published in Genepattern. SVM is a supervised classification method that finds the hyperplane in the data space that maximally divides the c classes. Given a random sample with data in that space, it will use this hyperplane to predict the class of the random sample. A confidence score is associated with each call.
Once the SVM has predicted the removed sample's class, the pipeline has finished the first run of the loop. It sequentially completes the loop M times, where M is the original population's sample size. After each sample has been removed and predicted once, the prediction result is compiled and is comprised of the percent correct calls and percent incorrect calls. Each sample is predicted with a certain level of confidence, and, based on prior experience, we have set an arbitrary threshold of confidence score >0.3 for a prediction to be recorded. If this score is <0.3, confidence is deemed to be insufficient, and the algorithm records a ‘no call’.
Results
Identifying peripheral blood cell populations whose frequency is altered in MS
In the screening phase of the analysis, we compared the frequency of peripheral blood cell populations found in 23 untreated RRMS subjects to that found in 17 healthy control subjects. The frequencies of 1018 such cell populations were compared in this assessment. We term each such frequency measurement in a distinct cell population a ‘frequency feature’, and 1018 represents the number of the frequency features that met our QC criteria. Of these frequency features, 123 have a permutation test P-value <0.05 in this screening analysis. To identify those features that may be truly differentiated between untreated RRMS subjects and healthy control subjects, we performed an extension analysis: we added data generated from 15 additional cases of untreated RRMS and 15 matched healthy control subjects to our analysis. Table 2 lists those frequency features for which (i) evidence for association with MS is enhanced after the addition of the extension phase subjects and (ii) the permuted P-value <0.05 in this extension analysis.
In reviewing the 22 features for which the evidence of association is enhanced in the extension analysis, the feature with the lowest permutation test P-value is the proportion of lymphocytes that are ‘CD8lowCD4’ (P = 0.0002); that is, on average, subjects with MS in our sample set have a smaller proportion of lymphocytes (experimental Gate 1, Supplementary Fig. 1) that express low levels of CD8 and do not express CD4 (Table 2). Figure 2A illustrates the gate used to define this CD8low population, a cluster of cells that is distinct from the CD8+ population. The distribution of our cytometric data for the CD8lowCD4− feature in subjects with MS and healthy control subjects is shown in Fig. 2B. Interestingly, this CD8low population is the one that appears to be present at a significantly higher frequency in healthy subjects than in subjects with MS in 20 of the 22 selected features in our screen. These 20 features are simply independent measurements of the entire CD8low population in different antibody pools located in different wells of the experimental plates. In fact, the four independently measured CD8lowCD4− features in our data set are all found within the 10 features with the lowest P-values. In merging the information provided by these 20 features, this cell population can be defined as being negative for CCR1, CCR2, CCR5, CCR7, CD14, CD27 and CxCR2 expression in various other antibody pools. However, antibody pool ‘a1’ (Table 2 and Supplementary Table 1a) provides a positive association: the CD8lowCD56+ cell population is reduced in RRMS subjects (Table 2).
In a secondary analysis, we carefully re-acquired the frequency of the different subsets of CD8lowCD4− cells in pool ‘a1’ by projecting these cells onto the CD3 and CD56 dimensions of staining (Supplementary Fig. 3). Most (mean of 53%) of these cells fall into a CD8lowCD4−CD56+CD3− cell population, and we find that its frequency is reduced in untreated subjects with RRMS (P = 0.02) (Supplementary Figs 3 and 4A). The smaller populations of CD8lowCD4−CD56+CD3+ cells and CD8lowCD4−CD56−CD3+cells are not significantly different when healthy and untreated RRMS subjects are compared (Supplementary Fig. 3). The CD8lowCD56+CD3−CD4− profile is consistent with the profile of a natural killer (NK) cell population (Robertson and Ritz, 1990).
Two other distinct lymphocyte populations captured in Gate 1 (‘lymphocyte gate’) (Supplementary Fig. 1) also appear in the features selected after the extension analysis (Table 2). Both are CD4+ cell populations. First, the proportion of CD4+CXCR2− cells in Gate 1 is greater in subjects with MS than in healthy control subjects (P = 0.004); this is probably the same cell population captured by the CD4+CD19− feature given the similarity in the frequencies of these two features in our healthy and MS subjects (Table 2). The distribution of frequencies for these two features amongst our subjects is presented in Supplementary Fig. 4. These populations capture the majority of CD4+ lymphocytes (Supplementary Fig. 2A). Given the screen's results, we re-acquired the frequency data for the different cell populations defined using anti-CD3, -CD4, and -CD8 antibodies and confirm that, aside from the CD8low cell population, only the CD4+ cell population displayed a modest difference in frequency when healthy subjects are compared to untreated RRMS subjects (Supplementary Fig. 5).
Replication of the CD8low cell population finding
Since the CD8lowCD4− cell population was identified as being reduced in frequency in untreated subjects with RRMS by multiple independent measurements, we analysed the same feature in data generated from an independent study, the CLIMB. These data were collected from a set of 16 untreated RRMS and 18 healthy control subjects that were not part of the MS Registry study, which generated the data used in our screen. The CLIMB data were also generated from blood samples ex vivo; the major distinction between the two studies is that, in CLIMB, PBMCs are extracted using a Ficoll gradient prior to staining. After processing the CLIMB data using our cytometric data gating protocol, we had four features that independently measured the CD8lowCD4− population in different antibody pools. The mean of these four features is reported in Table 3 and is consistent with the results of our extension analysis of the MS Registry data: in both cases, the proportion of CD8lowCD4− cells in the lymphocyte gate is ∼5% in subjects with MS and ∼7.5% in healthy control subjects (Tables 2 and 3). In this replication analysis using CLIMB data, we therefore validate our observation that the CD8low cell population is underrepresented in untreated RRMS subjects when these are compared to healthy control subjects (P = 0.0016).
Independent replication of the CD8low association to MS in CLIMB subjects
| CLIMB study data | |||
|---|---|---|---|
| Cell subset | Wilcox exact P-value | Healthy mean | Untreated RRMS mean |
| Gate 1/CD8lowCD4neg | 0.0016 | 7.50 | 5.10 |
| CLIMB study data | |||
|---|---|---|---|
| Cell subset | Wilcox exact P-value | Healthy mean | Untreated RRMS mean |
| Gate 1/CD8lowCD4neg | 0.0016 | 7.50 | 5.10 |
Independent replication of the CD8low association to MS in CLIMB subjects
| CLIMB study data | |||
|---|---|---|---|
| Cell subset | Wilcox exact P-value | Healthy mean | Untreated RRMS mean |
| Gate 1/CD8lowCD4neg | 0.0016 | 7.50 | 5.10 |
| CLIMB study data | |||
|---|---|---|---|
| Cell subset | Wilcox exact P-value | Healthy mean | Untreated RRMS mean |
| Gate 1/CD8lowCD4neg | 0.0016 | 7.50 | 5.10 |
The frequency of CD8low cells is also reduced in CIS
The MS Registry also collected the frequency of the CD8lowCD4− cell population in 38 subjects with CIS. When these subjects that have experienced a single clinical manifestation of inflammatory demyelination are compared to our 32 healthy control subjects in a secondary analysis, we observe a reduced frequency of CD8lowCD4− cells amongst the CIS subjects (P = 0.0006)(Fig. 2B). The mean frequency of this cell population is 5.6% in CIS subjects and 8.1% in healthy control subjects. The frequency of the CD8lowCD4− cell population in CIS subjects is similar to that seen in the MS subjects (P = 0.79) (Table 2). Therefore, CIS and RRMS subjects share at least one disturbance in cell population frequency, suggesting the existence of a shared pathophysiological process between these isolated and remitting–relapsing inflammatory processes.
Population structure in subjects with untreated RRMS or CIS
Clinical and pathological observations have long suggested the existence of subsets of MS subjects that have distinct disease courses and manifestations (Lucchinetti et al. 2001; Hauser and Oksenberg, 2006); however, clinical data alone have had limited success in identifying these subsets, especially early in the course of the disease. In an effort to discover such subject subsets, we explored the immunological architecture of our population of untreated subjects with RRMS. Using our MS Registry data set, we addressed this question empirically using a consensus clustering approach to estimate the number of subject clusters that comprise our sample of untreated RRMS subjects. This analysis returns three subject clusters as being the number that best fits the data set containing all 38 MS subjects with a complete immunological profile (screening and extension phase samples) (Fig. 3A and B); we obtain the same result when a non-redundant set of features (n = 795 out of the 1018 features of the complete profile) is used (Supplementary Fig. 6). The heatmap presented in Fig. 4 highlights the differences in expression that discriminate these three groups from each other. The features that best define each subset of subjects are listed in Supplementary Table 3. When the healthy control subjects are clustered into three subsets using the same methodology, a different set of distinguishing features is selected (data not shown).
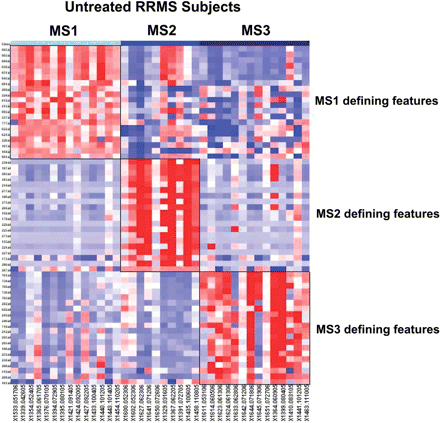
A heatmap highlights the difference in expression patterns between the three subsets of subjects with RRMS. In this heatmap, each column is an individual subject, with subjects grouped together based on the MS subset (MS1–3) to which they have been assigned by consensus clustering. Each row is a single feature. For each MS subset, the 20 features that are most differentiated in that subset were selected for inclusion in this heatmap. The exact nature of each feature is listed in Supplementary Table 3. Each cell is coloured along a gradient with red denoting high relative expression and blue low expression.
In looking at these lists of features, one must be careful in ascribing too much weight to the importance of individual features selected in this analysis since our subject sample remains relatively modest. With this caveat in mind, we see that the MS1 subset is the one that is distinguished by the frequency of CD8low and CD8 cell populations, while the other two classes of subjects appear to be defined by changes in the frequency of cells within our third gate (large, very granular cells) (MS2 subset) or in the frequency of CD14+ cell populations (MS3 subset) (Supplementary Table 3).
Examination of clinical data related to these subjects with MS is limited by our small sample size and revealed no clinical phenotype that is significantly correlated to one of the three subsets of subjects with untreated RRMS (Supplementary Table 4). The only suggestive result is the lower mean disease duration of subjects in the MS1 subset (P = 0.016). All samples were collected after a routine clinical visit, which occurs every 6 months for most patients at the Partners MS Center. None of these untreated RRMS subjects reported new symptoms or displayed clinical evidence of disease activity at these visits, and thus clinically evident bouts of CNS inflammation do not contribute to the population structure observed in our sample of subjects.
CIS subjects share at least one altered immunological feature, the proportion of cells that are CD8low, with RRMS subjects (Fig. 2B). However, a feature by feature analysis may often miss a broader pattern of modest differences, and so, to explore the relationship of CIS and RRMS immunological profiles more formally, we repeated the consensus clustering process described above on a pooled sample set containing all CIS and MS cases. This analysis finds that the 11 subjects with CIS that have a complete immunophenotypic profile are distributed proportionally among the three RRMS subgroups (Fig. 3C), suggesting that the population structure of subjects with RRMS reflects differences in immunological profiles that are present at the earliest stages of inflammatory demyelination, in cases of CIS.
Predicting a diagnosis of MS
While our primary analysis has found frequency features that are different between healthy and untreated RRMS subjects, these differences are not sufficient, on an individual feature basis, to distinguish these two subject categories. As seen with the CD8lowCD4− feature in Fig. 2B, the range of frequencies observed in healthy and untreated RRMS subjects overlap considerably. However, many modest differences can be powerful in discriminating subjects of two distinct classes, and we therefore implemented a tailored version of a cross-validation SVM to predict healthy control subjects from untreated RRMS subjects. We could not effectively segregate healthy control subjects from the RRMS subjects if the latter are pooled together into one phenotypic class (data not shown). However, once we acknowledge the MS population structure, the results of our prediction improved. In our small data set, the MS2 subgroup was most clearly distinguished from the pooled control samples (100% specificity, 94% sensitivity), and the MS1 and MS3 subgroups also displayed improved but still only modestly successful predictions over the pooled sample of subjects with MS (Supplementary Table 5). Thus, the panel of markers used in our analysis is not optimal for use in a diagnostic tool, but it does highlight the need to address MS population structure when designing future experiments with different panels of markers.
Discussion
We have applied rigorous statistical methods to unbiased analyses of cytometric data from healthy subjects and subjects with inflammatory demyelinating diseases; these analyses implicate one cell population in this family of diseases and uncover the architecture of our population of untreated subjects with RRMS or CIS. Specifically, we have validated the role of the CD8low cell population that is reduced to the same extent in CIS and RRMS subjects when they are compared to healthy control subjects. We also present evidence suggesting that these two categories of demyelinating disease share a similar population structure: both CIS and RRMS subjects appear to be distributed amongst three distinct clusters of subjects defined by different immunological profiles.
The reduced proportion of CD8low cells in subjects with MS was discovered by multiple independent assessments in our initial screen of whole blood stained for cytometric analysis. This finding was then confirmed by both (i) an extension of the original analysis and (ii) replication in an independent set of data generated from subjects in CLIMB. Furthermore, the same observation is made when healthy control subjects are compared to subjects with CIS. Thus, this decrease in CD8low cells is an early event in demyelinating diseases and is not an artefact of the way cells were stained in the MS Registry project. The observed difference, while statistically robust, is modest and is not sufficient to serve as a biomarker by itself (Fig. 2B). Nonetheless, because it can be effectively captured using two common antibodies (anti-CD4 and anti-CD8) that are used routinely in clinical laboratories, it could become a relatively simple and valuable component of diagnostic algorithm containing other clinical and radiological information for use by neurologists. In the future, as part of a broader diagnostic algorithm, this biomarker may be particularly useful to study individuals ‘at risk’ of developing MS (such as first degree relatives), if a reduction in this cell population predates the onset of the disease process.
The primary goal of the MS Registry, biomarker discovery, was therefore successfully accomplished; a measurement of the frequency of the CD8lowCD4− cell population in peripheral blood shows promise in possible future clinical application. However, this result is also important in what it reveals about the immunology of demyelinating diseases: it targets the CD8lowCD4− cell population for future exploration. In particular, it is intriguing that the same CD8lowCD4− cell population was found to be increased in frequency after treatment with daclizumab (anti-IL2Ra) in a recent Phase 2 trial (Bielekova et al., 2006). In this daclizumab trial, the expansion of the CD8low cell population after treatment correlated with decreased brain inflammation and decreased survival of activated T cells. Thus, correction of the deficit in CD8low cells that we find to be robustly associated with untreated RRMS and CIS subjects may be an important and early target for treatment in demyelinating diseases.
In our secondary analysis, we demonstrate that it is the CD56+CD3− subset of CD8lowCD4− cells that appears to drive the observation of reduced frequency in this population; in the daclizumab trial, it is also this CD8low CD56+CD4−CD3− subset of cells that appears to increase in frequency in response to treatment. This combination of markers suggests that these are NK cells that may have regulatory properties (Freud and Caligiuri, 2006). Further investigation will now be targeted at better characterizing the phenotype and function of these CD8lowCD4− cells to see which subset of NK cells may be implicated and how dysfunctional they may be in subjects with a demyelinating disease. Using different marker combinations, many investigators have assessed the role of NK cells in MS, and, while some early studies were negative, the propensity of the evidence available to date suggests that NK cell frequency is reduced in MS and that they may be dysfunctional (Segal, 2007). Comparisons of our data with these other studies are challenging at this point given the limited NK markers that we had in our panel and the heterogeneity of NK cell populations. Nonetheless, a CD56bright NK cell population has also been reported to be elevated in frequency during the last trimester of pregnancy, a time of reduced MS relapses, in women with MS (Airas et al., 2008). Similarly, an increased proportion of circulating CD56bright NK cells has been noted in subjects with RRMS following treatment initiation with IFN-β (Saraste et al., 2007). Finally, in vitro data suggest that CD56+ NK cells may help to regulate the activation of MBP-reactive T cells from subjects with (Takahashi et al., 2004). These small studies reinforce the suggestion that the frequency of CD56+ NK cells may have a role in MS. Thus, our novel description of a robust association between reduced CD8lowCD4− cell population frequency and a diagnosis of RRMS or CIS may be mediated at least in part by a deficit in CD56+ NK suppressive function that increases the likelihood of an autoimmune reaction.
Looking beyond the CD8low cell population, similarities between CIS and RRMS may extend to broader phenotypic profiles defined by our cytometric data; the underlying population structure identified by our consensus clustering method may be similar among CIS and RRMS subjects. The three subsets of subjects observed in both sets of samples suggest that population structure in inflammatory demyelinating diseases may be related to very early events in the pathophysiology of central nervous system inflammation: different triggers and/or immune dysfunction that occur early may eventually produce similar clinical manifestations that we define as RRMS. Since none of the included subjects displayed clinical manifestations of CIS or RRMS at the time of sampling, the subsets of subjects described here do not appear to be related to clinically evident episodes of inflammation.
The consensus clustering analysis that we present here suggests that collecting large immunological profiles may be one method with which to classify subjects with demyelinating diseases. However, independent replication of this observation is needed; further experimental work in larger sets of samples is required both to validate this approach and to select the optimal array of markers to be included in the profile. Our sample size, while substantial for this form of data, remains relatively small to powerfully explore the question of which cell populations are critical in defining each MS subset. In particular, technology and costs limit the number or different markers and marker combinations that we can assess: only 50 different antigens were assessed in 55 combinations of four antibodies in this project. Thus, while we have uncovered evidence of population structure in MS, we have not defined the key markers of each subset. In addition, our best estimate, based on our data, is that three major subsets of subjects exist in our data set, but much larger data sets will be more accurate in estimating the full distribution of subject subsets and in perhaps revealing rarer subsets. Such large studies would also refine the analysis of clinical variables that may be associated with different subsets of subjects. In general, immunological profiling appears to be one platform that will contribute significantly to the process of biomarker development, a process that must eventually integrate other forms of data such as imaging and genetic data in the development of effective diagnostic and prognostic models for MS and CIS.
In summary, our analyses direct us in three different directions: (i) to the exploration the use of the CD8low cell population in diagnostic and prognostic algorithms, (ii) to more detailed phenotypic categorization of CD8low cells in CIS and RRMS and (iii) to the validation and refined characterization of population structure in subjects with demyelinating diseases. Our data suggest that the next phase of studies must be much larger to powerfully assess this population structure, must target subjects early in their disease when they may have a clearer immunological profile, and must also contain new markers and new combinations of markers so that we may refine the cytometric signature of each subject subset.
Acknowledgements
We would like to thank our patients with MS and CIS that generously contributed blood samples to this project. We would also like to recognize the contribution of Lynn Stazzone as well as Drs Guy Buckle, Tanuja Chitnis, Susan Gauthier and Maria Houtchens in supporting subject recruitment and clinical data collection. P.L.D.J. is supported by NINDS K08 grant NS46341. The MS Registry data were produced as part of a collaboration between the Partners MS Center and Millenium Pharmaceuticals, Inc.
References
Abbreviations:
- CIS
clinically isolated demyelination; syndrome;
- CLIMB
Comprehensive Longitudinal Investigation of MS at the Brigham & Women's Hospital
- CMS
comparative marker selection
- FACS
fluorescence activated cell sorter
- GMFI
geometric mean fluorescence intensity
- MCV
mean channel value
- MedFI
median fluorescence intensity
- MFI
mean fluorescence intensity
- MRI
magnetic resonance imaging
- MS
multiple sclerosis
- NK
natural killer
- NMF
non-negative matrix factorization
- PBMCs
peripheral blood mononuclear cells
- QC
quality control
- RRMS
relapsing–remitting multiple sclerosis
- SVM
support vector machine
Author notes
*These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}