




Abstract
Synaptic inhibition is a central factor in the fine tuning of neuronal activity in the central nervous system. Symptoms consistent with reduced inhibition such as stiffness, spasms and anxiety occur in paraneoplastic stiff person syndrome with autoantibodies against the intracellular synaptic protein amphiphysin. Here we show that intrathecal application of purified anti-amphiphysin immunoglobulin G antibodies induces stiff person syndrome-like symptoms in rats, including stiffness and muscle spasms. Using in vivo recordings of Hoffmann reflexes and dorsal root potentials, we identified reduced presynaptic GABAergic inhibition as an underlying mechanism. Anti-amphiphysin immunoglobulin G was internalized into neurons by an epitope-specific mechanism and colocalized in vivo with presynaptic vesicular proteins, as shown by stimulation emission depletion microscopy. Neurons from amphiphysin deficient mice that did not internalize the immunoglobulin provided additional evidence of the specificity in antibody uptake. GABAergic synapses appeared more vulnerable than glutamatergic synapses to defective endocytosis induced by anti-amphiphysin immunoglobulin G, as shown by increased clustering of the endocytic protein AP180 and by defective loading of FM 1–43, a styryl dye used to label cell membranes. Incubation of cultured neurons with anti-amphiphysin immunoglobulin G reduced basal and stimulated release of γ-aminobutyric acid substantially more than that of glutamate. By whole-cell patch-clamp analysis of GABAergic inhibitory transmission in hippocampus granule cells we showed a faster, activity-dependent decrease of the amplitude of evoked inhibitory postsynaptic currents in brain slices treated with antibodies against amphiphysin. We suggest that these findings may explain the pathophysiology of the core signs of stiff person syndrome at the molecular level and show that autoantibodies can alter the function of inhibitory synapses in vivo upon binding to an intraneuronal key protein by disturbing vesicular endocytosis.
Introduction
Synaptic inhibition is a key-coordinative force in the CNS that is mediated predominantly by gamma-aminobutyric acid (GABA)-ergic and glycinergic interneurons. While the pathogenesis of some inherited and toxin-mediated diseases involving disturbed synaptic inhibition is well understood (Shiang et al., 1993; Lalli et al., 2003), it is unclear whether an antibody-mediated autoimmune process may also specifically affect synaptic inhibition. The pathogenic role of antibodies to ion channel-associated transmitter receptors has long been demonstrated in disorders of neuromuscular transmission (Toyka et al., 1975; Buchwald et al., 1998a, b, 2005). Furthermore, antibodies against membrane proteins, e.g. glutamate receptors, can alter excitatory transmission in the CNS (Sillevis Smitt et al., 2000; Coesmans et al., 2003; Dalmau et al., 2008).
Paraneoplastic stiff person syndrome (SPS) associated with antibodies to amphiphysin (Folli et al., 1993) is one of the few autoimmune diseases of the CNS with evidence for a direct pathogenic effect of autoantibodies. Clinical improvement correlates with lowering of antibody titres by therapeutic plasmapheresis (Wessig et al., 2003) and symptoms can be reproduced in rats by systemic passive transfer of patient immunoglobulin G (IgG) containing high-titre antibodies to amphiphysin (Sommer et al., 2005). Mechanistically, the typical core symptoms of SPS, i.e. stiffness, intermittent muscle spasms and anxiety (Murinson and Vincent, 2001; Meinck and Thompson, 2002; Vasconcelos and Dalakas, 2003), and the therapeutic effects of benzodiazepines are consistent with a disorder of GABAergic inhibition.
Amphiphysin is an intracellular protein involved in the synaptic vesicle cycle that promotes cleavage of clathrin-coated vesicles via binding of its Src homology 3 (SH3)—domain to dynamin (Wigge and McMahon, 1998). Acute blocking of the function of amphiphysin impairs synaptic vesicle endocytosis in vitro, leading to stimulation and frequency-dependent alteration of the presynaptic architecture with an increased number of clathrin coat intermediates and a decrease in the releasable vesicle pool. This results in a functionally relevant synaptic transmission failure, particularly at higher frequencies (Shupliakov et al., 1997; Evergren et al., 2004). In knockout mice with amphiphysin 1 deficiency, stimulus-dependent vesicle recycling is reduced, resulting in learning deficits and an increased susceptibility to seizures, consistent with reduced CNS inhibition (Di Paolo et al., 2002).
We describe a combined immunological, neurophysiological and molecular approach to investigating the specific pathogenic role of IgG antibodies toward amphiphysin and elucidating the disease mechanism. From our findings we propose a disorder affecting predominantly, but not exclusively, GABAergic inhibition.
Materials and methods
Patients and therapeutic plasma exchange
Two SPS patients with antibodies against amphiphysin suffered from a paraneoplastic SPS related to breast cancer. The clinical details of Patient 1 who had a complex paraneoplastic syndrome have been reported (Wessig et al., 2003). Patient 2 is a 52-year-old housewife whose initial symptoms included painful stiffness and spasms in the right arm. Symptoms deteriorated over time, resulting in impaired limb movements, stiff gait and eventually ataxia and cranial nerve pathology. Plasma exchange therapy (Cobe Spectra Apheresis System, CaridianBCT, Lakewood, Colorado, USA) involving roughly one plasma volume per session resulted in transient improvement of stiffness and muscle spasms. Titres of anti-amphiphysin antibodies were measured by means of a commercial enzyme immunodot assay with rabbit antisera raised against recombinant amphiphysin I as a positive control (H.P. Seelig, Karlsruhe, Germany). Titres before the first plasma exchange were 1–2 × 108 in Patient 1 and 1–5 × 105 in Patient 2, with a typical fall in titres upon plasma exchange.
Amphiphysin expression and IgG preparation
For expression and purification of recombinant human glutathione S-transferase-amphiphysin and glutathione S-transferase-SH3 domain fusion protein, the gene encoding for amphiphysin protein and the construct containing its wild-type SH3 domain (Grabs et al., 1997) were sub-cloned into the pGEX-6P expression vector system (GE Healthcare, Munich, Germany) to generate a glutathione S-transferase fusion protein (GE Healthcare) which was transformed into Escherichia coli BL21(DE3)-pRILP cells (Stratagene, La Jolla, CA, USA). The fusion proteins were expressed and purified as described earlier (David et al., 1994) and were cleaved by PreScission protease and separated from the glutathione S-transferase-tag according to the manufacturer’s instructions. For the generation of columns to use in amphiphysin affinity chromatography, amphiphysin protein was coupled to Cyanogen bromide-activated-Sepharose (GE Healthcare) as described by the manufacturer. Human immunoglobulins were purified from patient plasma filtrates as described earlier (Buchwald et al., 1998b; Sommer et al., 2005), including a control patient with chronic inflammatory polyneuropathy. For depletion of amphiphysin antibodies by affinity chromatography, purified IgG from Patient 1 that contained high titres of amphiphysin antibodies was dissolved in phosphate buffered saline and loaded onto an amphiphysin-sepharose affinity column. After washing, the amphiphysin-specific antibodies were eluted from the matrix with 0.1 M glycine, pH 2.5. Serial fractions of 2 ml each were collected and adjusted to pH 7.4 with 1 M Tris–HCl, pH 9.0. The amphiphysin antibody-depleted fractions and the amphiphysin antibody containing eluates were then dialysed separately against distilled water, freeze dried and stored at −20°C. Lyophilized IgG was dissolved in normal saline just before use and checked by western blotting.
Animals and surgery
Female Lewis rats were purchased from Harlan-Winkelmann (Borchen, Germany). All experiments were approved by the Bavarian State authorities. Intrathecal catheters (0.28 mm inner diameter; 0.61 mm outer diameter; intrathecal length: 7.0 cm; Supplementary Fig. 3A) were placed as described previously (Yaksh and Rudy, 1976). Rats that developed signs of paralysis after catheter placement were sacrificed immediately. After allowing recovery for 5 days, animals were randomized to receive either purified patient IgG with the highest titre of anti-amphiphysin antibodies without further processing (native-amphAB from Patients 1 and 2 at a concentration of 100 mg/ml, n = 10 and 6, respectively) or an IgG fraction that had been depleted of anti-amphiphysin antibodies by affinity chromatography (dep-amphAB, 100 mg/ml, n = 4, from Patient 1 only), or the specific anti-amphiphysin antibodies re-eluted from the affinity columns (spec-amphAB, diluted 1:10, concentration 10 mg/ml, n = 11, from Patient 1 only), the IgG fraction of plasmapheresis material from a control patient (control IgG, 100 mg/ml, n = 9 and n = 10 in two different experiments), and normal saline (n = 7). Intrathecal injections of 10 µl of patient IgG or saline with a subsequent flush of 10 µl saline were performed daily for 5 days, every other day for the next five injections and every third day for two following injections.
In parts of experiments we used amphiphysin knockout mice and their wild-type counterpart (kindly provided by P. deCamilli, Yale University, New Haven).
Behavioural analysis
Behavioural analyses were done by an investigator blinded to the treatment allocation of the rats. Animals were observed daily for at least 1 h while in their cages and when moving freely over a table with tunnels and obstacles. Each animal was videotaped for 3 min (Supplementary Videos) for documentation and blinded analysis. Stiffness was rated semi-quantitatively, similar to the clinical testing performed in humans with SPS (Dalakas et al., 2000). The following parameters were documented as abnormal signs on a scale of 0–3: slowing when in free motion, trunk stiffness during walking, reduced climbing ability, muscle spasms and dystonic movements (Fig. 1B). RotaRod testing and gait analysis were performed as described in Supplementary Methods.
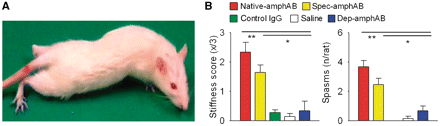
SPS-like symptoms after intrathecal passive transfer of anti-amphiphysin antibodies. (A and B) Rats treated with native-amphAB (n = 8) and purified spec-amphAB (n = 10), but none with control IgG (n = 9), or saline (n = 7) developed muscle stiffness and spasms with extension of the hind limbs and contraction of abdominal muscles. IgG fractions specifically depleted of the anti-amphiphysin antibodies (dep-amphAB, n = 4) had none of these effects. *P < 0.05; **P < 0.01.
Hoffmann reflex testing
Rats were anesthetized with intraperitoneal injections of ketamin and xylazin (80–100 mg/kg and 5 mg/kg, respectively). Hoffmann reflex recordings were performed as described earlier (Lee et al., 2005) using a Toennies electromyograph and NeuroScreenplus software (Erich Jaeger GmbH, Hoechberg, Germany). For analysis of post-activation depression, the ratio of the Hoffmann reflex and motor response (H/M ratio) was determined with serial single stimuli applied at the tibial nerve at frequencies ranging from 0.1 to 10 Hz with stimulation currents 30% above that needed for a maximal response (supramaximal) (Lee et al., 2005). H/M values of the tenth stimulation were taken for further analysis. The H/M ratio was fitted with an exponential decay: a = a0 + a1*exp(–f/f1), where a is the amplitude in percent (normalized to 100% at 0.1 Hz stimulation frequency) and f is the frequency in hertz (a0 + a1 were determined as 100%). The analysis of the D1/D2 inhibition is described in Supplementary Methods.
Dorsal root potential recording
Dorsal root potentials were recorded from dorsal roots L4 and L5 bilaterally with an ELC-03X amplifier (NPI Electronic, Tamm, Germany) after stimulation of the tibial nerve using a Grass S88 stimulator (stimulus duration 0.2 ms, single stimulation and trains of three stimuli at 100 Hz; Grass Technologies, West Warwick, Rhode Island, USA) as described earlier (Barron and Matthews, 1938; Schmidt, 1971). In a subset of animals, the dependence of the dorsal root potential amplitude on GABAergic transmission was tested by applying bicucullin locally to the dorsal root entry zone of the spinal cord.
Histological and immunohistochemical analysis
At the end of the experiment, rats were sacrificed, blood was withdrawn and the lumbar spinal cord was rapidly removed, mounted in Tissue-Tec optimal-cutting-temperature embedding compound and then deep frozen in liquid nitrogen. For details on histological and immunohistochemical procedures, see Supplementary Methods.
Stimulation emission depletion microscopy
Ten-micrometer spinal cord sections or dissociated hippocampal neurons were viewed with a Leica SP5 true confocal scanner stimulation emission depletion microscope (Hell, 2007) using a 100× oil immersion lens (Leica Microsystems, Wetzlar, Germany) with the following filter and wavelength settings: for visualization of Cy-3 secondary antibodies excitation, 543 nm and emission 575–620 nm; for stimulation emission depletion imaging with the Atto-conjugated secondary antibody excitation, 635 nm and emission 645–715 nm.
Imaging with quantum dot-coupled antibodies
Three hundred microlitre spec-amphAB and control IgG antibodies (1 mg/mg) were coupled to the activated quantum dots according to manufacturer’s instructions, resulting in an IgG concentration of 344 ± 3.6 µg/ml. For live cell imaging, 2.5 µl of each tagged antibody solution was dissolved in 200 µl cell culture medium and incubated with dissociated hippocampal neurons cultured from wild-type and amphiphysin knockout mice for 10 min, 1.5, 3 and 6 h, respectively. For detection of internalized antibodies, nanocrystal-tagged antibodies (130 nmol) were incubated with the cell cultures for 6 h at 37°C. After fixation, cells were incubated overnight at 4°C with antibodies against calnexin, a protein of the endoplasmic reticulum (Abcam, Cambridge, UK), followed by Cy2 secondary antibodies (1:100) and 4’,6-diamidino-2-phenylindole (DAPI) nuclear staining. Stacks of confocal images were recorded in the multi-track mode and examples of single optical slices are shown in Fig. 5A. Images were analysed with Image Pro Plus version 4.5 software (MediaCybernetics, Bethesda, Maryland, USA).
Antibody internalization assays
To reveal epitope specificity, a defined amount (46 nmol) of quantum dot-tagged spec-amphAB was pre-incubated with recombinant amphiphysin in concentrations from 388 nmol to 0.8 fmol in the cell culture medium at room temperature for 1 h followed by incubation with dissociated mouse hippocampal neurons for 6 h at 37°C. For each concentration, five randomly distributed square fields of 375 µm side length were analysed by counting internalized quantum dot profiles and overall number of neurons (mean count of neurons was 92.2 ± 6.4 per profile). Means of internalized quantum dot counts per profile were plotted against the antigen concentration using Sigma-Plot 9.0 Software (Systat Software, San Jose, CA, USA). Other hippocampal neurons were incubated with 46 nmol tagged spec-amphAB and unlabelled spec-amphAB in various concentrations (ranging from 1.8 µmol to 0.34 nmol) for 6 h at 37°C, and then processed and analysed as described earlier. To investigate the temperature dependence of the internalization process, hippocampal neurons were incubated at 37°C or 25°C for 6 h in additional experiments. Mean numbers of internalized quantum dots were plotted against the concentration of unlabelled spec-amphAB. All experiments were performed in duplicate.
Analysis of defects in vesicle endocytosis
To analyse the influence of anti-amphiphysin antibodies on vesicle recycling and endocytic function in GABAergic and glutamatergic synapses, dissociated hippocampal neurons were incubated with native-amphAB and control IgG at 100 µg/ml for 6 h at 37°C. In a separate set of experiments, cell cultures were additionally incubated with 1 µmol of tetrodotoxin (TTX, Sigma, St. Louis, Missouri, USA). Neurons were then fixed with ice-cold methanol (100%) for 20 min at −20°C and subsequently washed three times with phosphate buffered saline. For confocal imaging, cells were incubated at 4°C with the following antibodies: mouse monoclonal anti-bassoon antibodies (Stressgen, 1:1000), rabbit polyclonal anti-AP180 antibodies (Synaptic Systems, 1:2000) followed by the respective Cy-3 conjugated secondary antibodies (Dianova, 1:300) for 2 h at room temperature and DAPI nuclear staining. For high-resolution stimulation emission depletion microscopy, double immunofluorescence staining was performed with anti-AP180 and polyclonal guinea pig anti-vesicular glutamate transporter (VGLUT) or anti-vesicular GABA transporter (VGAT) antibodies (both Synaptic Systems, Goettingen, Germany, 1:2000, 1:1000, respectively) followed by anti-guinea-pig Cy-3 secondary antibodies and anti-rabbit IgG conjugated to Atto 647N (Atto-Tec, Siegen, Germany). Clustering of the synaptic proteins was analysed by counting immunoreactive puncta per 300 µm2 in six randomly chosen areas in each of six single-culture incubations according to previous reports (Ferguson et al., 2007; Hayashi et al., 2008). Colocalization with VGAT and VGLUT was determined in each of 10 randomly chosen areas using Image Pro Plus version 4.5 software.
High-performance liquid chromatography analysis of GABA and glutamate release
Hippocampal cell cultures were incubated with native-amphAB and control IgG as described earlier. After 6 h, medium was removed and neurons were pre-incubated for 90 s at 37°C with 200 µl of 119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM Hepes and 30 mM glucose (artificial CSF). Thereafter, cell cultures were either stimulated by depolarization with 200 µl of a high-potassium solution (31.5 mM NaCl, 90 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM Hepes and 30 mM glucose) or 200 µl artificial CSF for 90 s at 37°C (Ferguson et al., 2007; Hayashi et al., 2008) (n = 6 for each condition). GABA and glutamate concentrations were analysed by high-performance liquid chromatography as described in the Supplementary Methods. To assess structural integrity, subsets of cells were stained with rabbit anti-β-tubulin antibodies (Sigma, 1:1000) followed by anti-rabbit Cy-3 secondary antibodies.
Analysis of vesicle endo- and exocytosis with FM dye imaging
Dissociated hippocampal neurons were grown on glass cover-slips with a grid (Science Services, Munich, Germany) until Day 10 in vitro and were then incubated for 3 h with all IgG fractions at a concentration of 100 µg/ml. Thereafter, cells were superfused with artificial CSF. Synaptic boutons were loaded with 10 µM FM® dye 1–43 (Molecular Probes, Eugene, Oregon, USA) in the presence of 90 mM KCl and 2 mM CaCl2 for 90 s at room temperature according to published protocols (Klingauf et al. 1998). Individual boutons were imaged after 15 min perfusion with artificial CSF. Destaining of the synaptic boutons was performed with brief pulses of 90 mM KCl and 2 mM CaCl2. Boutons were viewed with a Zeiss Axioskope 2 microscope using a 63× water immersion lens (Zeiss, Wetzlar, Germany). FM dyes were excited at 470 nm wavelength with a shutter-controlled X-Cite® 120PC fluorescence lamp (Exfo Life Sciences Division, Ontario, Canada) every 2 s and images were taken with an Axiocam MRm camera (Zeiss). After imaging, a bright-field image was taken for later identification of the depicted neurons and cells were fixed with ice-cold 100% methanol as described. Cells were then stained for VGAT and VGLUT using Cy-3 and Cy-2 secondary antibodies, respectively. The pixel intensities of fluorescent spots in the live-imaging episode were analysed using Image Pro Plus version 4.5 software. GABAergic and glutamatergic boutons were identified after the live-imaging experiments by comparison with the VGAT and VGLUT staining, respectively (Fig. 7B).
Whole-cell patch-clamp analysis
GABAergic miniature potentials and single evoked GABAergic inhibitory postsynaptic currents were recorded in granule cells of acute hippocampal slice preparations after pre-incubation with 1 mg/ml native-amphAB or control-IgG at 32°C for at least 30 min. For further details see Supplementary Methods.
Primary cell cultures
Rat embryonic spinal neuron co-cultures (motor neurons and interneurons), mouse embryonic motor neuron cultures and mouse embryonic hippocampal cultures were prepared as described in Supplementary Methods.
Data analysis
All data are presented as means and standard errors of the means. Statistical analyses were performed using SPSS software version 11.5 (SPSS Inc., Munich, Germany) with ANOVA and Bonferroni post hoc test for the behavioural analyses, analysis of Hoffmann reflex data (Supplementary Fig. 6), dorsal root potentials and immunohistochemical quantifications. The FM dye evoked fluorescence intensity was assessed for each time point, and the intergroup differences were analysed with a one-way ANOVA with Bonferroni post hoc analysis. Hoffmann reflex data (Fig. 3A; Supplementary Fig. 2) and in vitro electrophysiological data (mean inhibitory postsynaptic current in steps of 10 stimuli throughout the train and mean inhibitory postsynaptic current in the recovery phase) were analysed using the Mann–Whitney U-test.
Results
Intrathecal passive transfer of anti-amphiphysin antibodies to the spinal cord induces long-lasting SPS-like symptoms in rats
To investigate the functional impact of amphiphysin autoantibodies, we used native-amphAB which was obtained from plasma filtrates of Patient 1 in passive transfer experiments. Following this, we performed experiments with specific antibodies to amphiphysin purified by affinity chromatography from Patient 1 IgG, with this patient’s IgG specifically depleted of amphiphysin antibodies with a control patient’s IgG and with the IgG fraction of Patient 2, who had anti-amphiphysin antibody positive SPS but with lower titres. We repetitively applied intrathecal injections of these IgG fractions to the subarachnoid space of rats via implanted spinal catheters. After six or seven injections of IgG containing anti-amphiphysin antibodies, all rats treated with native-amphAB (8/8 from Patient 1 and 6/6 from Patient 2) and with affinity-purified spec-amphAB (10/10 from Patient 1) developed stiffness of trunk and limb muscles with intermittent muscle spasms and gait abnormalities (Fig. 1A and B; Supplementary Figs 1 and 2A and Supplementary Videos 1–3). In contrast, none of the four rats injected with the same amounts of IgG depleted of specific antibodies (dep-amphAB) and none of the 19 rats injected with control IgG developed SPS-like symptoms. Very rarely, animals of the control groups showed short-lasting truncal stretching without twisting, possibly due to transient local irritation of the spinal cord by the catheters (Supplementary Fig. 3). Footprint analysis showed increased outward rotation of the hind paws in rats treated with native-amphAB and spec-amphAB (Supplementary Fig. 1A and B). The grip force test of the forelimbs did not reveal differences between groups (data not shown), indicating no detectable effects of anti-amphiphysin antibodies on forelimb muscle strength. On the accelerating RotaRod, rats in the native-amphAB group showed a marked and significant decrease in forced walking time (Supplementary Fig. 1C and Supplementary Video 2).
Purified human anti-amphiphysin antibodies bind specifically to neuronal structures and recognize the amphiphysin SH3 domain
The various IgG preparations of Patient 1 used in the passive transfer experiments were tested by western blotting with homogenized rat spinal cord and with a 45 kDa glutathione S-transferase fusion protein containing the amphiphysin SH3 domain, the high-affinity binding partner for dynamin (Shupliakov et al., 1997; Evergren et al., 2004). The native-amphAB and spec-amphAB showed clear and strong bands, equivalent to blots with polyclonal rabbit anti-amphiphysin antibodies. In the depleted fraction (dep-amphAB), only a faint band at 128 kDa was present, equivalent to a purification efficiency of ∼99.9% (Fig. 2A and B).
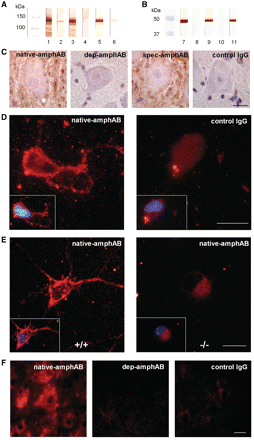
Selective binding of anti-amphiphysin antibodies to neuronal tissue. Western blotting of purified IgG (1 mg/ml) on rat spinal cord tissue (A) and on a glutathione S-transferase SH3 domain fusion protein (B) displayed specific binding of anti-amphiphysin antibodies (lanes 1 and 7: native-amphAB IgG; 3 and 9: spec-amphAB from Patient 1; 4 and 10: control IgG; 5 and 11: commercial, polyclonal anti-amphiphysin antibodies). Affinity chromatography resulted in almost complete depletion of anti-amphiphysin antibodies (lanes 2 and 8: dep-amphAB; 6: native-amphAB at 1000× lower concentration). (C) Naïve rat spinal cord sections were incubated with IgG (n = 3 for each IgG preparation). Incubation with native-amphAB and with spec-amphAB from Patient 1 resulted in distinct staining of neuronal plasma membranes and dendrites, indicating specific binding to neuronal surface structures. After depletion of anti-amphiphysin antibodies these staining properties were completely absent. Control IgG at the same concentration showed no immunoreactivity (scale bar: 30 µm). (D) Incubation of a rat spinal motor neuron-interneuron co-culture with native-amphAB IgG and subsequent immunoreaction against human IgG antibodies resulted in an intense punctate staining of the cell membrane, soma and dendrites, whereas control IgG elicited only faint staining of cell bodies (representative sample out of three for each IgG preparation; scale bar: 20 µm). Inset shows overlay of the same neurons with DAPI). (E) Incubation of wild-type (+/+) mouse motor neurons with native-amphAB revealed a similar staining pattern as in D (left panel), whereas motor neurons from amphiphysin knockout mice (–/–) did not (scale bar: 20 µm). (F) Treatment of rats with pathogenic native-amphAB, but not with dep-amphAB or control IgG, resulted in intense staining of spinal motor neurons in lumbar spinal cord sections (scale bar: 30 µm).
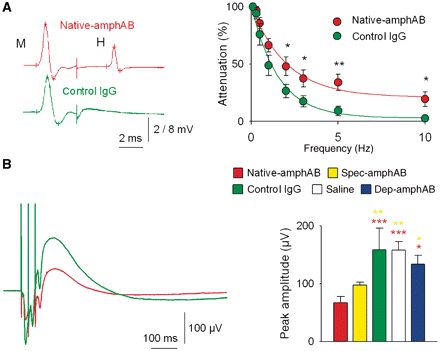
Reduced GABAergic spinal inhibition. (A) Left: in Hoffmann reflex recordings at 10 Hz stimulation, native-amphAB treated rats (n = 8) reached a plateau of H-wave amplitudes at 21% of the initial amplitude in contrast to the almost complete (3%) amplitude reduction in the control IgG group (n = 9). Right: representative Hoffmann reflex recordings with native-amphAB (upper trace) and with control IgG. (B) GABAergic dorsal root potentials evoked after a train of three stimuli (100 Hz) were significantly lowered in the native-amphAB (n = 10, red asterisks) and spec-amphAB groups (n = 17, yellow asterisks) compared with the control IgG (n = 15) and saline groups (n = 12) and rats treated with IgG depleted of anti-amphiphysin antibodies (n = 17) (*P < 0.05; **P < 0.01; ***P < 0.001).
In spinal cord sections from untreated rats, only incubation with native-amphAB and spec-amphAB but not with dep-amphAB and control IgG resulted in perineuronal staining in the ventral horn (Fig. 2C). When primary motor neuron and interneuron co-cultures were immunoreacted with the IgG fractions at a 10 µg/ml concentration, only native-amphAB but not control IgG resulted in a strong-sprinkled-staining pattern of neuronal cell bodies and dendrites (Fig. 2D). To further substantiate the specificity of binding to amphiphysin, we switched species and incubated motor neurons or spinal cord sections of wild-type mice with the IgG preparations. An identical staining pattern was observed as in rat tissue, whereas cells and sections from amphiphysin knockout mice showed only faint background staining (Fig. 2E; Supplementary Fig. 4).
Human IgG was detected in the homogenized spinal cord of rats that had been injected intrathecally with native-amphAB (120 µl at a concentration of 100 mg/ml resulting in 135 ± 23 µg/ml tissue, n = 3) and in the group injected with specific antibodies (spec-amphAB, 120 µl at a concentration of 10 mg/ml resulting in 67 ± 9 µg/ml tissue, n = 4), indicating relatively higher binding of spec-amphAB and thus pointing to a possible enrichment at the respective binding sites. Plasma levels of human IgG in the rats were usually below the detection threshold, which made a systemic effect of the administered antibodies unlikely (data not shown).
Immunofluorescence staining for human IgG showed intense immunoreactivity of neurons in the ventral horn and in the centromedullary region in spinal cord cryosections from rats treated in vivo with the pathogenic native-amphAB or spec-amphAB but not with control IgG (Fig. 2F). Taken together, these findings suggest specific binding of anti-amphiphysin antibodies to neuronal structures containing amphiphysin.
Anti-amphiphysin antibodies disturb spinal synaptic pathways by reduction of GABAergic presynaptic inhibition
The abnormal continuous motor activity and hyperexcitability in monosynaptic reflexes (Floeter et al., 1998) suggest disordered spinal inhibitory mechanisms in SPS. To assess this in our rat model, we investigated spinal inhibition in antibody treated animals in vivo. We examined an established monosynaptic pathway by measuring changes of Hoffmann reflex amplitude (the electrically elicited deep tendon reflex). Hoffmann reflexes are normally inhibited during high-frequency stimulation, which is due to post-activation depression at the afferent neuron-motor neuron synapse (Lev-Tov and Pinco, 1992; Lee et al., 2005) (Supplementary Fig. 5A). Indeed, in animals treated with control IgG (n = 9 and 10 in separate experiments), frequency-dependent depression of the Hoffmann reflex was observed as expected. However, after application of native-amphAB or spec-amphAB, post-activation depression of the Hoffmann reflex amplitude was significantly diminished. This effect was not seen in rats injected with dep-amphAB (Fig. 3A and Supplementary Fig. 2B).
Another pathophysiologically relevant feature associated with the typical stiffness of SPS is incomplete inhibition of antagonistic muscles leading to co-activation of agonistic and antagonistic muscles. Therefore, we investigated the modulation of the Hoffmann reflex amplitude by applying a conditioning volley with different latencies at a nerve supplying an antagonistic muscle (Eccles et al., 1961; Lee et al., 2005; Chen et al., 2006) (Supplementary Fig. 5B). No depression in Hoffmann reflex was found in the rats treated with the pathogenic native-amphAB (n = 8) and spec-amphAB (n = 7) (for details see Supplementary Fig. 6).
The reduction in frequency-dependent depression of Hoffmann reflex amplitudes and depression after antagonistic stimulation may also involve polysynaptic pathways. Therefore we examined presynaptic inhibition, one of the most potent spinal inhibitory mechanisms (Eccles et al., 1962a, b, 1963; Hultborn, 2006) mediated by depolarization of primary afferent presynaptic terminals through selective GABAergic axo-axonal synapses of inhibitory interneurons (termed ‘last order primary afferent depolarization’ interneurons). Dorsal root potentials are electrotonically conducted potentials generated by primary afferent depolarization interneurons that can be recorded from lumbar dorsal roots in vivo (Barron and Matthews, 1938; Eccles et al., 1963; Hultborn, 2006) (Supplementary Fig. 5C and D). Dorsal root potentials showed a characteristic uniform course and could be blocked by local superfusion of bicucullin (De Koninck and Henry, 1994) (Supplementary Fig. 7A and C). After single and repetitive stimulation, dorsal root potential amplitudes were significantly reduced in rats treated with any of the anti-amphiphysin antibody fractions as compared with animals treated with control IgG (Fig. 3B; Supplementary Fig. 2C, 7B and Table 1), which is best explained by a marked reduction of presynaptic GABAergic inhibition in animals treated with pathogenic anti-amphiphysin antibodies. However, a possible influence of a decreased excitatory feed-forward input to spinal inhibitory interneurons cannot be excluded. These effects on GABAergic inhibition were not seen when IgG depleted of the specific antibodies (dep-amphAB) was used. Of note, the effects on dorsal root potential amplitudes and on Hoffmann reflex depression were less pronounced when native-amphAB from Patient 2 was used. This might be explained by the lower antibody titres (105 versus 108) of these fractions.
Patient anti-amphiphysin antibodies are specifically internalized in vivo and in vitro and bind to presynaptic nerve terminals
To address the question of whether the amphiphysin-specific antibodies enter neurons, the site of antibody binding and internalization at the sub-cellular level was studied. Spinal cord sections from rats treated with native-amphAB and spec-amphAB were double immunoreacted with antibodies to anti-human IgG and to presynaptic antigens including VGAT, VGLUT, bassoon and clathrin. High-resolution stimulation emission depletion light microscopy (Kittel et al., 2006; Hell, 2007) revealed staining in punctuate structures around motor neurons and dendrites. Human IgG showed close colocalization with the presynaptic vesicle-associated VGAT, partial colocalization with VGLUT and clathrin, and a juxtaposed but not overlapping distribution with the presynaptic active zone protein bassoon, suggesting accumulation in presynaptic terminals at the sites of the vesicle cycle (Fig. 4).
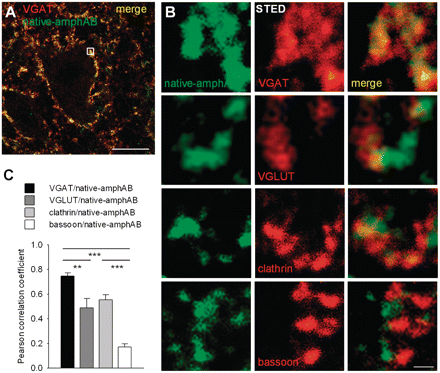
Presynaptic localization of intrathecally injected anti-amphiphysin antibodies. (A) Double immunofluorescence labelling anti-human IgG and the presynaptic markers VGAT, showed punctate staining around motor neurons and dendrites in the anterior horn of the spinal cord (scale bar: 20 µm). Staining for VGLUT, clathrin and bassoon gave similar patterns. (B) Stimulation emission depletion high-resolution microscopy from an area of interest (small square in the left panel) revealed almost complete overlay of human IgG with VGAT, partial overlay with VGLUT and clathrin, and largely adjacent staining with the active zone marker bassoon, indicating presynaptic enrichment of injected anti-amphiphysin antibodies (scale bar: 500 nm). (C) The amount of colocalization was determined using volumetric data and calculating Pearson’s correlation coefficient (**P < 0.01; ***P < 0.001).
In contrast to autoantibodies directed at membrane-bound receptors (Toyka et al., 1975; Lang et al., 1983; Sillevis Smitt et al., 2000; Coesmans et al., 2003), it has been discussed controversially whether antibodies to an intracellular antigen can be pathogenic, and if so, how they reach their target antigen (Kissel and Elble, 1998; Levin et al., 2002). To further study binding and internalization, we incubated dissociated mouse hippocampal neurons with spec-amphAB tagged with fluorescent nanocrystals. We observed a strong and time-dependent binding followed by internalization of the antibody complexes (Supplementary Fig. 8). After incubation of wild-type neurons with tagged control IgG or after incubation of amphiphysin knockout neurons with spec-amphAB, only scarce if any binding was observed. Intracellular binding of tagged anti-amphiphysin antibodies was verified with confocal microscopy and colocalization with the intracellular marker calnexin (linked to the endoplasmic reticulum) (Fig. 5A). Pre-incubation of tagged spec-amphAB at increasing concentrations of recombinant amphiphysin inhibited the internalization of antibodies in a concentration-dependent manner (Fig. 5B), suggesting an epitope-specific process. Internalization of labelled antibodies was inhibited with a calculated EC50 of 15.7 nmol by adding unlabelled spec-amphAB in increasing concentrations to a constant amount of tagged antibodies (57.5 nmol) (Fig. 5C). Reducing temperature led to a marked reduction of internalization (Fig. 5D), consistent with a temperature-dependent mechanism of internalization, e.g. clathrin-mediated endocytosis (Smith et al., 2008).
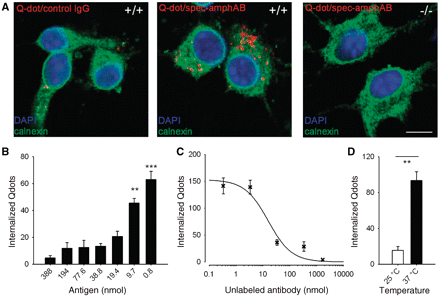
Specific neuronal uptake of anti-amphiphysin antibodies. (A) Confocal microscopy of dissociated hippocampal neurons incubated with quantum dot (Qdot) and stained with the intracellular marker calnexin showed internalization of anti-amphiphysin antibodies in wild-type neurons (+/+), but only minimal neuronal uptake of tagged control IgG. No specific internalization of anti-amphiphysin antibodies was observed with neurons from amphiphysin knockout mice (−/−) (scale bar: 10 µm). (B) Preabsorption with the antigen by pre-incubation of tagged spec-amphAB with recombinant amphiphysin resulted in a concentration dependent decrease of free binding sites of the antibodies and lead to diminished uptake of anti-amphiphysin antibodies (saturated conditions from 38.8 nmol). (C) Pre-incubation with tagged spec-amphAB (46 nmol) and various amounts of unlabelled spec-amphAB revealed a decrease of internalized antibodies with increasing amounts of unlabelled antibodies, confirming the epitope-specific binding and subsequent uptake of anti-amphiphysin antibodies. Fitting a Hill equation, Q = Qmax × EC50/(EC50 + Ab) resulted in an EC50 of 15.7 nmol (Q, number of internalized Qdots; Ab, concentration of unlabelled antibody). (D) Reducing temperature resulted in a marked decrease of internalized tagged spec-amphAB (**P < 0.01; ***P < 0.001) indicating a temperature-sensitive uptake mechanism.
Endocytic function of inhibitory synapses is preferentially affected by anti-amphiphysin antibodies
The clinical signs of SPS and our experimental results reported above are in line with defective GABAergic inhibition. Since amphiphysin is expressed in most CNS synapses, and not just in GABAergic presynaptic terminals, we set out to investigate the effects of patient IgG on inhibitory and excitatory synapses directly. In dissociated hippocampal neurons, incubation with native-amphAB but not with control IgG resulted in an increased clustering of the endocytic protein AP180 (Fig. 6A and B), which is known to result from defective endocytosis (Ferguson et al., 2007; Hayashi et al., 2008). Blocking the continuous neuronal background activity with TTX led to a redistribution of AP180 clustering, indicating a significant endocytic defect already at basal network activity. Stimulation emission depletion microscopy revealed a significantly increased clustering of AP180 at GABAergic synapses, marked with VGAT, as compared with glutamatergic synapses, marked with VGLUT (Fig. 6C and D). When measuring the neurotransmitter release in cell culture supernatants with high-performance liquid chromatography, incubation with native-amphAB resulted in a significant decrease of neurotransmitter release at basal levels and even more so after stimulation with 90 mmol KCl (Fig. 6E and F). Thereby the GABA release (78% compared with incubation with control IgG) and glutamate release (53% of control IgG) were both decreased. Notably, after incubation with anti-amphiphysin antibodies, brief stimulation with 90 mmol KCl no longer resulted in a significant increase in GABA release. The structural integrity of dissociated hippocampal neurons after high-potassium stimulation was ascertained by visual inspection after staining against β-tubulin (Supplementary Fig. 9).
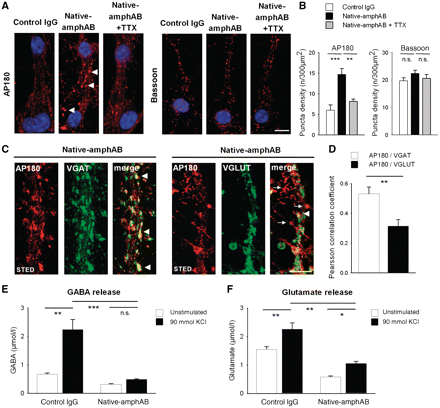
Functional effects of autoantibodies on GABAergic and glutamatergic synapses. (A and B) Confocal imaging of mouse hippocampal neurons treated either with native-amphAB or control IgG revealed enhanced clustering (arrowheads) of the endocytic protein AP180, but not of the active-zone protein bassoon in neurons pre-incubated with anti-amphiphysin antibodies as compared with control IgG. AP180 was redistributed in cultures when synaptic network activity was blocked by tetrodotoxin (TTX; **P < 0.01; scale bar 10 µm). (C and D) Clustering of the endocytic protein AP180 occurs preferentially at GABAergic synapses. High-resolution stimulation emission depletion microscopy revealed significantly more overlay of GABAergic synapses (stained with VGAT) with clusters of AP180 (arrowheads), as compared with glutamatergic synapses (stained with VGLUT) (arrows; scale bar 5 µm; **P < 0.01). (E and F) Following incubation with native-amphAB high-performance liquid chromatography analysis of cell culture supernatant revealed a substantial decrease of GABA release (down to 21.8% compared with incubation with control IgG) and, to a lesser extent, of glutamate release (down to 46.6%; P < 0.01). Stimulation with 90 mM K+ in anti-amphiphysin IgG treated cell cultures did not result in an increase of GABA release, but did increase glutamate release. This indicates a more pronounced alteration of inhibitory as compared with excitatory synaptic activity (*P < 0.05; **P < 0.01; ***P < 0.001).
Next we investigated the functional effect of anti-amphiphysin antibodies on the vesicle cycle. When synaptic boutons of dissociated hippocampal neurons were loaded with FM dyes (Fig. 7A and B), subsequent destaining with 90 mmol KCl pulses was faster after pre-incubation with native-amphAB and spec-amphAB as compared with dep-amphAB and control IgG (τ = 14, 12, 25 and 48 s, respectively), whereby the first phase of rapid destaining was preferentially affected (Fig. 7C). Since loading of synaptic boutons with FM dye–stained vesicles depends on effective endocytosis, the faster destaining of boutons by anti-amphiphysin antibodies may be explained by defective endocytosis leading to fewer releasable vesicles. We then compared the FM dye destaining kinetics separately in GABAergic and glutamatergic synapses (Fig. 7B). Treatment with spec-amphAB but not with dep-amphAB led to a faster destaining also in glutamatergic synapses (τ = 19 versus 51 s). The loading capacity of GABAergic boutons was markedly reduced to 59% as compared with glutamatergic boutons within the same experiments (Fig. 7D), corroborating a substantial defect in vesicular endocytosis, preferentially in GABAergic synapses.
![Vesicle endocytosis and subsequent transmitter release is preferentially affected in GABAergic synapses. (A and B) Synaptic boutons of dissociated hippocampal neurons were loaded with FM dye 1–43. Destaining by vesicle exocytosis was elicited using brief stimulation with 90 mM K+ and images were taken every 2 s. GABAergic (circles) and glutamatergic (squares) synapses were identified after the live imaging by VGAT und VGLUT immunofluorescence [inset in (A); scale bar: 20 µm (A), 50 µm (B)]. (C) Analysis revealed a faster-destaining time course of synapses treated with native-amphAB (n = 3 experiments; 116 boutons analysed) or spec-amphAB (n = 4; 133 boutons) compared with dep-amphAB (n = 4; 134 boutons) or control IgG (n = 4; 164 boutons) (P < 0.05). (D) Differentiating between GABA- and glutamatergic synapses-revealed slightly faster destaining of glutamatergic synapses after pretreatment with spec-amphAB (n = 3; 61 boutons) compared with dep-amphAB (n = 3; 63 boutons) (P < 0.01, 15–18 s after the start of destaining). However, in GABAergic synapses (n = 3; 66 boutons), the initial FM dye loading intensity was markedly reduced (P < 0.001) compared with glutamatergic synapses (within the same experiments) and to both kinds of synapses pre-treated with dep-amphAB (n = 3, 64 boutons for GABAergic synapses). This indicates insufficient endocytosis resulting in a diminished number of releasable vesicles.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/133/11/10.1093_brain_awq253/1/m_awq253f7.gif?Expires=1716404061&Signature=rco7ymVmicWTgBWFyvL8ZeCB6axHOg3lPj2wYrbc4au-5cZCO4ZrCjztdBoToXMpcbVAMYsMuNwefUzKmp0ul5QS0pJjF~VZa0VH0LvpvujYr1ttkZZi0m-dOs-zOCO0QuugkfIhUnPv7KYKEHxqJBhNocIy~klyo~oPcADcTzosqD4t1q2NBH3ydDWHbogkwP-3i6XShoxUV9pdTfKFcRxOwpTt8myh0qkLV~K4iNqd71E5a0QGuI6Lo5Ck5qzajk8cz2TuMVKqAUmeeCXgvDJwJQmuR33mRjupIkrfPOQQaapMYoa-XMvqFLZ~TQAZAyX0Ero9MD1kNfTOKHXbgg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Vesicle endocytosis and subsequent transmitter release is preferentially affected in GABAergic synapses. (A and B) Synaptic boutons of dissociated hippocampal neurons were loaded with FM dye 1–43. Destaining by vesicle exocytosis was elicited using brief stimulation with 90 mM K+ and images were taken every 2 s. GABAergic (circles) and glutamatergic (squares) synapses were identified after the live imaging by VGAT und VGLUT immunofluorescence [inset in (A); scale bar: 20 µm (A), 50 µm (B)]. (C) Analysis revealed a faster-destaining time course of synapses treated with native-amphAB (n = 3 experiments; 116 boutons analysed) or spec-amphAB (n = 4; 133 boutons) compared with dep-amphAB (n = 4; 134 boutons) or control IgG (n = 4; 164 boutons) (P < 0.05). (D) Differentiating between GABA- and glutamatergic synapses-revealed slightly faster destaining of glutamatergic synapses after pretreatment with spec-amphAB (n = 3; 61 boutons) compared with dep-amphAB (n = 3; 63 boutons) (P < 0.01, 15–18 s after the start of destaining). However, in GABAergic synapses (n = 3; 66 boutons), the initial FM dye loading intensity was markedly reduced (P < 0.001) compared with glutamatergic synapses (within the same experiments) and to both kinds of synapses pre-treated with dep-amphAB (n = 3, 64 boutons for GABAergic synapses). This indicates insufficient endocytosis resulting in a diminished number of releasable vesicles.
Since studying GABAergic transmission in spinal cord slices requires differentiation between monosynaptic and polysynaptic input (Torsney and MacDermott, 2006), we turned to a well-defined, simple model synapse which is capable of high-frequency synaptic transmission with known high turnover of presynaptic vesicles (Kraushaar and Jonas, 2000). We performed whole-cell patch-clamp analysis of a monosynaptic GABAergic inhibitory transmission on hippocampal granule cells in acute slice preparations after incubation with native-amphAB and control IgG (Fig. 8A; Supplementary Fig. 10). The analysis of spontaneous GABAergic miniature potentials and of single evoked inhibitory postsynaptic currents revealed no difference between groups in amplitudes, rise time and decay time, indicating no relevant synaptic transmission defect at resting conditions. In contrast, when GABAergic afferents were stimulated at high frequencies, the amplitude of evoked inhibitory postsynaptic currents decreased faster in slices treated with antibodies against amphiphysin (τ = 0.9 versus 1.7 s), and it was significantly smaller throughout the first phase of train simulation. Additionally, pre-incubation with native-amphAB led to lower absolute amplitude of GABAergic inhibitory postsynaptic currents in the recovery phase (Fig. 8B).
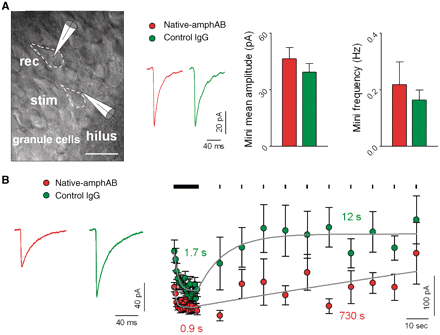
(A) Whole-cell recording of granule cells (cell margins marked by dashed lines) in the hippocampal dentate gyrus showed only minor and not significant differences in amplitude or frequency of GABAergic miniature inhibitory postsynaptic currents when slices were pre-incubated with native-amphAB or control IgG (examples are averaged traces from single experiments; stim, stimulating electrode; rec, recording electrode; scale bar: 20 µm). (B) Analysis of mean inhibitory postsynaptic currents amplitudes during high frequency stimulation (10 Hz) showed significantly lower amplitudes in native-amphAB treated slices during the first phase of the train response (P < 0.001 for stimuli 10–20, P < 0.05 for stimuli 20–30) and in the recovery period (P < 0.05; native-amphAB: n = 7, control IgG: n = 10). Traces show averaged responses from two single experiments during train stimulation. After high-frequency stimulation, GABAergic inhibitory postsynaptic currents decline faster in native-amphAB treated cells compared with control IgG (τ = 0.9 s versus 1.7 s) and also the recovery of inhibitory postsynaptic currents amplitudes is substantially slower after the train stimulation (τ = 730 s versus 12 s).
Discussion
The principal finding of our study is that human disease-related antibodies against the SH3 domain of amphiphysin, an intracellular protein involved in synaptic transmission, are capable of inducing a disorder of synaptic inhibition. We provide experimental evidence for the concept that these effects are brought about by binding of the antibodies to CNS neurons with subsequent internalization, in turn leading to a disturbance of synaptic vesicle endocytosis and, consequently, to diminished transmitter release.
The behavioural observations in rats treated with antibodies to amphiphysin from two patients with SPS were strikingly similar to the core signs of these and other SPS patients and are consistent with reduced synaptic inhibition.
Specificity of the antibody effects
We used several strategies to test the amphiphysin specificity of the observed effects: (i) the effects observed in vivo were strictly related to anti-amphiphysin antibodies; (ii) as shown with high-resolution stimulation emission depletion microscopy, injected antibodies accumulated in presynaptic terminals at the site of vesicle endocytosis, consistent with the known distribution of the target antigen and the anatomical site of the functional defects identified by electrophysiology; (iii) internalization of anti-amphiphysin antibodies into hippocampal neurons was dependent on the presence of amphiphysin, as shown by competition experiments. Corroborating this notion, no specific internalization could be observed in neurons from amphiphysin-deficient mice. Collectively, these findings make it unlikely that an additional antibody of unknown specificity within the patients’ polyclonal IgG fraction might have been responsible for the observed effects on synaptic inhibition.
The pathogenic role of circulating autoantibodies associated with diseases of the CNS has long been a matter of debate, unlike in autoimmune disorders of the neuromuscular junction (Toyka et al., 1975; Vincent et al., 1995; Buchwald et al., 1998a, 2005) and peripheral nerves (Fukunaga et al., 1983; Roberts et al., 1994; Buchwald et al., 1998a). In only few conditions, pathogenic effects of autoantibodies in CNS diseases were proposed (Rogers et al., 1994; Sillevis Smitt et al., 2000; Coesmans et al., 2003; Lennon et al., 2005) and very recently shown for the disorder neuromyelitis optica (Bennett et al., 2009; Bradl et al., 2009; Saadoun et al., 2010). In all these conditions, the crucial antigen is a receptor or ion channel on the cell membrane, or a protein adjacent or linked to such receptors or channels. In contrast, it has usually been considered unlikely that antibodies to an intracellular protein antigen might become pathogenic, and if they were, it remained unclear how they would reach their target antigen (Kissel and Elble, 1998; Levin et al., 2002). We here show data suggesting that patient antibodies can be internalized into neurons and colocalize with other intracellular proteins in vivo and in vitro by an epitope-specific process.
Deficient GABAergic inhibition as a pathophysiologic mechanism in SPS
Several lines of argumentation lead to the hypothesis of disturbed GABAergic inhibition in SPS. (i) Electromyography shows persistent motor activity in patients and in the animal model (Meinck et al., 1984; Sommer et al., 2005); (ii) patients usually respond to GABAergic drugs like benzodiazepines; (iii) all three other types of autoantibodies associated with SPS to date directly target GABAergic structures: glutamate decarboxylase (Solimena et al., 1988), GABA-receptor-associated protein (GABARAP) (Raju et al., 2006) and gephyrin (Butler et al., 2000). It is unclear why GABAergic synapses should be preferentially affected by anti-amphiphysin antibodies, since amphiphysin has a widespread distribution in the CNS with a preferential occurrence in presynaptic terminals around synaptic vesicles (Lichte et al., 1992; David et al., 1996). Recently, a striking heterogeneity in synaptic vesicle recycling and functional properties between inhibitory and excitatory synapses was shown in a mouse model of defective endocytosis due to dynamin 1 deficiency (Ferguson et al., 2007; Hayashi et al., 2008). Inhibitory GABAergic synapses were selectively affected by endocytic deficiency, which was attributed to their intrinsic properties, in particular to their higher level of tonic activity and spiking frequency with a fast synaptic vesicle turnover (Kraushaar and Jonas, 2000; Luthi et al., 2001).
To test spinal pathways in our experimental rats treated intrathecally with the purified patient IgG preparations, we used similar paradigms as were used previously by others in patients with SPS (Floeter et al., 1998). In animals injected with anti-amphiphysin antibodies, frequency-dependent Hofmann reflex depression, Hofmann reflex depression after antagonistic stimulation and dorsal root potential peak amplitudes were all significantly lowered, which is best explained by a frequency-dependent reduction in inhibitory spinal synaptic function. Taken together, this makes antibody mediated reduction of GABAergic inhibition a likely disease mechanism in anti-amphiphysin associated SPS. In support of these findings, our in vitro experiments showed that the endocytic and consecutively exocytic function of GABAergic synapses were clearly more affected than that of glutamatergic synapses upon incubation with antibodies against amphiphysin: (i) enhanced clustering of the endocytotic protein AP180; (ii) a decrease of neurotransmitter release and faster destaining and (iii) decreased initial vesicle staining of FM dyes are all consistent with a defect of endocytosis and a subsequent decrease in the releasable vesicle pool resulting in reduced synaptic transmission. Moreover, our patch-clamp analysis revealed a substantial decrease of GABAergic transmission during high-frequency stimulation and in the ensuing recovery phase, but not under resting conditions, which is again consistent with a decrease of the releasable vesicle pool after repetitive synaptic activity. This is in accordance with previous studies, where injection of experimentally generated anti-amphiphysin antibodies into the lamprey giant synapse led to a greatly reduced presynaptic vesicle pool and to an increased number of clathrin-coated intermediates (Shupliakov et al., 1997; Evergren et al., 2004). Accordingly, in amphiphysin-deficient mice, deficits in synaptic vesicle recycling were unmasked only by stimulation (Di Paolo et al., 2002), indicating a high degree of compensation of the altered amphiphysin function in vivo, similar to what we could observe in our passive-transfer animal model. Extrapolating these findings to SPS patients with anti-amphiphysin antibodies, we hypothesize that their GABAergic inhibition may be deficient in a state of high-excitatory activity, such as in stressful situations where finely tuned inhibitory drive is needed. This hypothesis would match the observed worsening of SPS symptoms with sudden noises or frightening emotions. Nevertheless, direct experimental comparison of the effects on excitatory and inhibitory synapses and tonic versus phasic synapses, by antibodies against amphiphysin, should be elucidated in future studies.
In conclusion, the observed phenotype and the electrophysiological alterations in the experimental rats in vivo and the in vitro findings collectively support our concept that a preferential disturbance of GABAergic inhibitory synaptic activity may be the crucial pathophysiological basis of SPS.
Funding
Deutsche Forschungsgemeinschaft SFB 581, (TP A7 and HE 2621/4-1); Interdisciplinary Clinical Research Centre (IZKF) of the University of Würzburg School of Medicine; Intramural University Research Funds.
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
We thank M. Sendtner for helpful suggestions and discussions and for critically reading the article and R.F. Schmidt for helpful advice and discussion of the electrophysiological recordings. We are indebted to P. De Camilli for his continuous support and encouragement, for providing the cDNA clone encoding for amphiphysin protein, the constructs containing its wild-type SH3 domain, amphiphysin KO mice and for helpful suggestions on the interpretation of our findings. L. Biko, S. Hellmig, B. Dekant, H. Bruenner, B. Broll, R. Burger and H. Wetzstein provided expert technical assistance in animal experiments, histology, high-performance liquid chromatography and IgG preparations. The authors declare no competing financial interests.
References
Abbreviations
- DAPI
4′,6-diamidino-2-phenylindole
- dep-amphAB
patient IgG depleted of anti-amphiphysin antibodies
- IgG
immunoglobulin G
- native-amphAB
native IgG fraction containing high-titre anti-amphiphysin antibodies
- spec-amphAB
affinity purified and reconstituted patient anti-amphiphysin antibodies
- SPS
stiff person syndrome
- VGAT
vesicular GABA transporter
- VGLUT
vesicular glutamate transporter
Author notes
*These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}