




Abstract
We describe here an innovative, non-transgenic animal model of Alzheimer's disease. This model mimics early stages of sporadic disease, which represents the vast majority of cases. The model was obtained by interfering with the complex between a disintegrin and metalloproteinase domain containing protein 10 (ADAM10), the main α-secretase candidate, and its partner, synapse-associated protein 97, a protein of the postsynaptic density-membrane associated guanylate kinase family. Association of ADAM10 with synapse-associated protein 97 governs enzyme trafficking and activity at synapses. Interfering with the ADAM10/synapse-associated protein 97 complex for 2 weeks by means of a cell-permeable peptide strategy is sufficient to shift the metabolism of the amyloid precursor protein towards amyloidogenesis and allows the reproduction of initial phases of sporadic Alzheimer's disease. After 2 weeks of treatment, we detected progressive Alzheimer's disease-like neuropathology, with an increase of β-amyloid aggregate production and of tau hyperphosphorylation, and a selective alteration of N-methyl-d-aspartic acid receptor subunit composition in the postsynaptic compartment of mouse brain. Behavioural and electrophysiological deficits were also induced by peptide treatment.
Introduction
Alzheimer's disease is the most common cause of dementia and no cure is available at the moment. The underlying neuropathology of Alzheimer's disease includes extracellular deposition of β-amyloid peptide (Aβ), intraneuronal accumulation of aberrant forms of hyperphosphorylated tau (Selkoe, 2001) as well as synapse dysfunction and neuronal loss (DeKosky and Scheff, 1990; Selkoe, 2002).
Substantial data indicate accumulation of Aβ in the brain, particularly in its non-fibrillar soluble oligomeric forms, as the primary influence driving Alzheimer's disease pathogenesis (Hardy and Higgins, 1992). The rest of the disease process, including formation of neurofibrillary tangles and synapse loss, is considered a downstream consequence of Aβ deposition and it is proposed to result from an imbalance between Aβ production and Aβ clearance. Aβ derives from the concerted action of β–secretase (Vassar et al., 1999), which mediates amyloid precursor protein shedding at Aβ N-terminus and γ-secretase, responsible for amyloid precursor protein C-terminal stub cleavage (De Strooper et al., 1998). Alternatively, amyloid precursor protein processing is exerted by a disintegrin and metalloproteinase (ADAM10) family member that prevents Aβ formation (Lammich et al., 1999; Postina et al., 2004).
The generation of animal models that recapitulate characteristic features of Alzheimer's disease is of great relevance to improve the understanding of disease pathogenesis and to develop new therapies. Current models of Alzheimer's disease rely on information gathered from inherited familiar forms. Identification in various human pedigrees of gene mutations in amyloid precursor protein (Goate et al., 1991), in presenilin-1 and -2 (Scheuner et al., 1996) provided a potential tool of generating rodent models of the disease. Despite their undoubted value, transgenic Alzheimer's disease mice show a number of pitfalls, i.e. they fail to model the progressive stages of the disease, which questions their role as pre-clinical models of Alzheimer's disease. There is no mouse line that models aetiology of late-onset/age-related sporadic Alzheimer's disease, which accounts for the vast majority of cases. Thus, focusing on new hypotheses concerning Alzheimer's disease pathogenesis and on the corresponding modelling is now a need.
Recently, Kim and colleagues reported two potentially pathogenic mutations with incomplete penetrance for late-onset familial Alzheimer's disease in the region encoding the prodomain sequence in the gene for ADAM10 (Kim et al., 2009). ADAM10 interacts directly with synapse-associated protein (SAP) 97, a cargo protein of the postsynaptic density-membrane associated guanylate kinase (PSD-MAGUK) family (Marcello et al., 2007). Since SAP97 is involved in the trafficking of molecules to the glutamatergic synapse (Leonard et al., 1998; Sans et al., 2001) and in events controlling synaptic transmission, its role in neurodegenerative diseases has been largely addressed and alterations have been identified in early stages of several disorders (Nash et al., 2005; Lau and Zukin, 2007). The direct binding of proline-rich domains of the tail of ADAM10 to the SH3 domain of SAP97 promotes ADAM10 trafficking to synaptic membranes and regulates enzyme activity on amyloid precursor protein in primary neuronal cultures and, acutely, in rodent brains (Marcello et al., 2007). Therefore, interfering with ADAM10/SAP97 complex appears as an ideal approach to investigate the connection between amyloid precursor protein metabolism and synaptic dysfunction in early phases of Alzheimer's disease.
Here, we describe the generation of an innovative mouse model of Alzheimer's disease through uncoupling of ADAM10 from SAP97. Disruption of the ADAM10/SAP97 complex is sufficient to reduce ADAM10 levels at synaptic sites as well as to decrease its activity, thus shifting amyloid precursor protein metabolism towards amyloidogenesis and reproducing the different events of sporadic Alzheimer's disease.
Material and methods
Ex vivo studies
Hippocampi from six patients with Alzheimer's disease and six healthy controls were obtained from The Netherland Brain Bank. Established Braak and Braak criteria were used to categorize Alzheimer's disease tissues (Braak and Braak, 1991). Healthy controls had no history of psychiatric or neurological disease and no evidence of age-related neurodegeneration. All procedures were in accordance with the National Institutes of Health Guide for Care and Use of laboratory human tissues and were approved by the Ethics Committee of the University of Milan.
Animal treatment
Adult male C57BL/6 mice, 8 weeks old, were treated with the cell-permeable peptide Tat-Pro ADAM10709-729 (Pro, NH2-YGRKKRRQRRRPKLPPPKPLPGTLKRRRPPQP-COOH), obtained by linking the 11 amino acid human immunodeficiency virus Tat transporter to a 21 amino acid sequence corresponding to the ADAM10 proline-rich domains, or with Tat-Ala ADAM10709-729 (Ala, NH2-YGRKKRRQRRRAKLAAAKALAGTLKRRRAAQA-COOH) (Xigenpharma, Lausanne, CH). Peptides were conjugated to fluorescein. Peptides were administered subcutaneously for 7 or 14 days through osmotic minipumps (Alzet2002, Charles River, Calco, LC, Italy) or injected intraperitoneally for 1 day. Dose regimens corresponded to 1 nmol/g. Animals were killed 1, 7 or 14 days after treatment and the brain was rapidly dissected. Four independent experiments with the two groups, Ala and Pro (three animals per group) were performed for biochemical experiments. Animal handling and surgical procedures were carried out with care taken to minimize discomfort and pain in accordance with ethical regulations and guidelines of the European Communities Council (Directive of 14 November 1986, 86/609/EEC).
Sample preparation
Triton-X insoluble fraction (TIF) and soluble fraction S2 were obtained as previously described (Gardoni et al., 2001; Marcello et al., 2007).
Sodium dodecyl sulphate polyacrylamide gel electrophoresis
Samples (homogenate and TIF) were separated by electrophoresis on a 7% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS–PAGE) gel, while the S2 fraction was separated on a 6% SDS–PAGE gel. To detect amyloid precursor protein carboxy terminal fragments (CTFs), Tris–Tricine gels at 15% were used. Proteins were transferred to a nitrocellulose membrane and probed with the corresponding primary antibody followed by incubation with appropriate horseradish peroxidase-conjugated secondary antibodies (Pierce, Rockford, IL, USA).
Immunohistochemistry
Mice were deeply anaesthetized with tribromoethanol (intraperitoneally, 1 mg/kg) and perfused through the ascending aorta with physiological saline followed by fixative solution (4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4). Brains were removed and post-fixed overnight at 4°C in the same fixative. Series of 50 µm thick coronal sections were cut using a vibratome. For Congo Red staining, slices were washed in water, incubated in 1% Congo Red solution and then differentiated in alkaline alcohol (0.2% KOH in 80% ethanol) and counterstained with haematoxylin. For tau phosphorylation immunostaining, sections were washed in phosphate-buffered saline, treated with 2% H2O2 in phosphate-buffered saline and incubated in 5% normal goat serum in phosphate-buffered saline containing 0.5% Triton X-100. Sections were incubated with anti-tau AT8 antibody at a concentration of 5 mg/ml, overnight at 4°C. Biotinylated goat-anti-mouse IgG (Vector, Burlingame, CA, USA) was applied at a dilution of 1:500, followed by incubation with streptavidin-conjugated horseradish peroxidase (Vector) for 1 h. Tissue-bound peroxidase was detected using 3,3-diamino-benzidine (0.025%) as a chromogen. Immunoreacted sections were placed on microscope slides and coverslipped with mounting medium.
Immunoprecipitation
Fifty μg of (for ADAM10/SAP97, Akap79/150/SAP97, GluR1/SAP97 and PYK-2/PSD-95 immunoprecipitation) or 70 µg (for SAP97/NR2A immunoprecipitation) of homogenate from mice treated with Ala or Pro were incubated overnight with an antibody against ADAM10, Akap79/150, GluR1, PYK2 or SAP97. Co-immunoprecipitation was carried out as previously described (Marcello et al., 2007).
Determination of Aβ release
For evaluation of Aβ release from mice treated with Ala or Pro, an enzyme-linked immunosorbent assay kit was used (IBL, Gunma, Japan). The test was performed on total homogenate following the manufacturer's instructions. A standard curve ranging from 0 to 800 pg/ml was obtained with each point corresponding to the average of two measurements.
Quantification and statistical analysis
Quantification of western blot analysis was performed by computer-assisted imaging (Quantity-One® System, BioRad, Hercules, CA, USA) and statistical evaluations were performed according to two-tailed paired Student's t-test or one-way ANOVA followed by appropriate post hoc tests. Quantification of immunostaining is shown as fold difference compared with controls (healthy controls or Ala) in the same experiment. Values are means ± SEM for four independent experiments for a total of 24 animals.
Hippocampal slice preparation and electrophysiology
Fourteen days after surgical minipump insertion, mice were decapitated without anaesthesia. Hippocampi were quickly dissected out in ice-cold oxygenated artificial CSF containing the following (in mM): 120 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 26.2 NaHCO3, 1.0 NaH2PO4 and 11.0 glucose, bubbled with a mixture of 95% O2/5% CO2, pH 7.4. Transverse hippocampal slices (400 µm; n = 8 slices from n = 3 mice per group) were cut and maintained in a humidified holding chamber at room temperature for 1 h for recovery. Individual slices were transferred to an interface recording chamber (Fine Science Tools, Foster City, CA, USA) where they were continuously perfused with artificial CSF maintained at 29°C, at a flow rate of 1.5–2 ml/min. Extracellular field excitatory postsynaptic potentials were recorded in the stratum radiatum of area CA1 of the hippocampus using glass electrodes filled with 3 M NaCl coupled to the input stage of a Warner IE-210 amplifier (Handen, CT, USA). Stimuli were delivered to the commissural/Schaffer collateral afferents at 0.033 Hz (0.1 ms pulse duration) with a concentric bipolar stainless steel electrode. Data were collected and analysed online (10 kHz sampling rate) using pClamp software (Clampex, Axon Instruments, CA, USA). A stimulus intensity that elicited an extracellular field excitatory postsynaptic potential amplitude that was ∼33% of the maximum was used. Long-term potentiation was induced by a high-frequency stimulation, consisting of four trains of 100 Hz for 1 s, separated by 30 s. Synaptic activity was measured as the maximal slope of the rising phase of the extracellular field excitatory postsynaptic potential. Each experiment was normalized to the mean value of the extracellular field excitatory postsynaptic potential slope recorded during the baseline (20 min of stable recording before high-frequency stimulation). Long-term potentiation values were calculated as the percentage increase in the extracellular field excitatory postsynaptic potential slope 50–60 min after the end of high-frequency stimulation compared with the baseline value. Data are presented as means ± SEM, and ANOVA was used for statistical analysis.
Novel object recognition
Twelve days after surgical minipump insertion, mice treated with Ala and Pro (n = 9 per group) were habituated to the test arena for 5 min in the absence of any object. In the first trial, the mice were presented for 10 min with two objects of identical size and shape. In the second trial, the mice were presented for 10 min with a duplicate of one of the objects from the first trial and a new object differing in shape. During both trials, exploration of the object was defined as the time spent sniffing, licking or touching the object. The discrimination index [novel (t) − familiar (t)]/[total time (t) at novel + familiar], number of total movements in 5 min and time (s) spent investigating in 10 min were evaluated. Results are shown as mean ± SEM, and ANOVA followed by Fisher as a post hoc test were used for statistical analysis.
DyOlistic labelling
Fourteen days after surgical minipump insertion, mice treated with cell-permeable peptides Pro and Ala (n = 6 per group) were anaesthetized and perfused with paraformaldehyde. Transverse hippocampal CA1 apical and cortical layer I slices (300 µm) were cut with a vibratome and slices were kept in phosphate buffer. Tungsten particles were coated with a lipophilic dye, DiI (Molecular Probes, Eugene, OR, USA). Microcarriers were loaded into the gene gun and then fired into tissue slices with the BioRad Gene Gun with a gas pressure 200 psi at a distance of 40 mm. Slices were kept for 10 min in 0.1 M phosphate buffer and then for 2 h in 4% paraformaldehyde. After 24 h, images of DiI-labelled structures in hippocampal CA1 stratum radiatum and layer I of the somatosensory cortex were acquired using the Zeiss Confocal LSM510 microscope with ×63 objective. At least 10 z-stack images were acquired for each animal and for each area of interest. The ImageJ software program was used to quantify DiI-labelled neurons. Dendritic segments were chosen randomly and were at least one soma's length away from the cell soma. Four parameters were measured, including spine density, spine area, spine length and spine head diameter. All measurements were made with the investigator blinded to the treatment used. Results are shown as means of n = 33 neurons ± SEM (at least 10 spines were measured for each neuron) and Student's t-test was used for statistical analysis.
Antibodies
The following antibodies were used: polyclonal antibody anti-ADAM10 and polyclonal antibody AB2072 against amyloid precursor protein from Abcam (Cambridge, MA, USA); monoclonal antibody anti-GluR1, monoclonal antibody anti-PYK-2 and monoclonal antibody anti-α-CaMKII from Millipore (Billerica, MA, USA); monoclonal antibody anti-SAP97 from Stressgen (Ann Arbor, MI, USA). Polyclonal antibody anti-Actin, monoclonal antibody anti-Tubulin and monoclonal antibody anti-NR2A from Sigma (St Louis, MO, USA); monoclonal antibody AT8 against tau from Pierce; monoclonal antibody anti-GluR2 and monoclonal antibody anti-PSD-95 from Neuromab (Davis, CA, USA); monoclonal antibody anti-NR1 and polyclonal antibody anti-NR2B from Invitrogen (Carlsbad, CA, USA); polyclonal antibody anti-AKAP79/150 from Santa Cruz Biotechology (Santa Cruz, CA, USA); and monoclonal antibody anti-N-Cadherin from BD Bioscience (Sparks, MD, USA).
Results
ADAM10/SAP97 association is reduced in mice after 2 weeks of treatment with Pro peptide and in Alzheimer's disease patients at Braak level 4
Interfering acutely with the ADAM10/SAP97 complex inhibits ADAM10 trafficking to the postsynaptic membrane and its activity in rodents (Marcello et al., 2007).
On the basis of this observation, we supposed that subchronic disruption of the ADAM10/SAP97 complex could mimic one of the events occurring in sporadic Alzheimer's disease and could allow the generation of a model of Alzheimer's disease.
To investigate this, Tat-Pro ADAM10709–729 peptide (Pro) (Marcello et al., 2007) was used, in which ADAM10 proline-rich domains (amino acids 709–729), responsible for binding to SAP97, are fused to the Tat transporter sequence (Aarts et al., 2002; Fig. 1A). Eight-week-old mice were exposed subcutaneously for 2 weeks to the Pro peptide. As a control, we used an inactive peptide, Tat-Ala ADAM10709-729 (Ala), where all prolines were substituted by alanines.
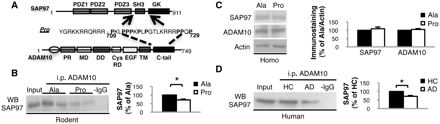
ADAM10/SAP97 association is reduced after 2 weeks of treatment with Pro peptide and in patients with Alzheimer's disease. (A) Schematic representation of SAP97, ADAM10 and Pro peptide. (B) C57BL/6 mice were treated for 2 weeks with Pro or Ala peptides and total brain homogenates were co-immunoprecipitated with anti-ADAM10 antibody. SAP97 presence was evaluated in the immunocomplex. Treatment with Pro peptide decreases the association of the two proteins (P = 0.005). (C) Western blot of homogenate from Pro- and Ala-treated mice performed with anti-ADAM10 and anti-SAP97 antibodies. No changes were detectable in ADAM10 and SAP97 levels. All data were normalized to actin. (D) Homogenate from hippocampi of healthy controls (HC) and patients with Alzheimer's disease (AD) was immunoprecipitated with anti-ADAM10 antibody and SAP97 co-precipitation was evaluated. ADAM10/SAP97 association is reduced in Alzheimer's disease patients compared with healthy controls (P = 0.001). For all experiments, quantitative analysis of immunostaining is shown as percentage of control (Ala or healthy controls) in the same experiment. i.p. = immunoprecipitation; WB = western blot.
To validate our tool, peptide penetration into neurons was confirmed by double staining with an anti-Tat antibody, used to visualize peptides, and an anti-MAP-2 antibody, used as a neuronal marker (Supplementary Fig. 1). Peptide capability to interfere with the association between SAP97 and ADAM10 was investigated by co-immunoprecipitation experiments from total brain homogenates. A reduction of the ADAM10/SAP97 immunocomplex was observed in mice treated with Pro compared with Ala (Fig. 1B; n = 4, P = 0.005, −41.3 ± 6.5%, Pro versus Ala). We found no modifications of SAP97 and ADAM10 levels in total homogenate, suggesting that the observed uncoupling can be ascribed to a decreased association rather than to changes in levels of these proteins (Fig. 1C; ADAM10: n = 4; P > 0.05; +12.6 ± 4.9%; SAP97: n = 4; P > 0.05; +9.7 ± 10.7%, Pro versus Ala).
To validate the relevance of the ADAM10/SAP97 complex for Alzheimer's disease pathogenesis, we performed a biochemical analysis of hippocampi from Alzheimer's disease patients at Braak level 4 (Braak and Braak, 1991) and of age-matched healthy controls. Braak level 4 corresponds to initial stages of sporadic disease, and the hippocampus was analysed as a strongly affected area. Total brain homogenates from hippocampi of healthy controls and patients with Alzheimer's disease were immunoprecipitated with anti-ADAM10 antibody, and SAP97 co-immunoprecipitation was evaluated. We revealed a decrease of the ADAM10/SAP97 immunocomplex in Alzheimer's disease patients compared with healthy controls (Fig. 1D; n = 6; P = 0.001, −33.7 ± 4.9%, Alzheimer's disease versus healthy controls), suggesting uncoupling of the ADAM10/SAP97 complex as an early event in Alzheimer's disease pathogenesis. No differences were detected between healthy controls and patients with Alzheimer's disease in SAP97 and ADAM10 levels in total brain homogenates (SAP97: n = 6, P > 0.05; +31.4 ± 35.1%; ADAM10: n = 6, P > 0.05, −7.7 ± 11.7%, Alzheimer's disease versus healthy controls), underlying that the ADAM10/SAP97 complex is not reduced in Alzheimer's disease due to lower expression of the two proteins.
Pro peptide is specific in disrupting ADAM10/SAP97 complex
We investigated Pro peptide specificity in disrupting the ADAM10/SAP97 complex, without influencing other protein–protein interactions.
First, the capability of SAP97 SH3 domain to bind other partners in the presence of Pro was evaluated. Cell-based studies suggest that Akap79/150, a signalling scaffold, is recruited to AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid) receptors through interaction with SH3/GK domains of SAP97 (Colledge et al., 2000). We performed co-immunoprecipitation experiments between SAP97 and Akap79/150. No alterations were found in Akap79/150/SAP97 association (Fig. 2A; n = 4, P > 0.05, −8.7 ± 31.3%, Pro versus Ala). SAP97 has been recognized as a cargo element responsible for synaptic trafficking of key elements of the glutamatergic synapse, i.e. the NR2A subunit of the glutamate N-methyl-d-aspartic acid (NMDA) receptor (Gardoni et al., 2003) and the GluR1 subunit of the glutamate AMPA receptor (Sans et al., 2001). Pro specificity was assessed by analysing the binding of these partners to SAP97 by co-immunoprecipitation between SAP97 and NR2A, as well as between SAP97 and GluR1. No alterations were found in either SAP97/NR2A association (Fig. 2B; n = 4, P > 0.05, +6.3 ± 20.4%, Pro versus Ala) or in the SAP97/GluR1 immunocomplex (Fig. 2C; n = 4, P > 0.05, −21.2 ± 9.4, Pro versus Ala) after treatment. Moreover, the Pro peptide did not interfere with binding of other synaptic SH3 to Pro-rich domains, as assessed by co-immunoprecipitation of PYK2 and PSD-95 (Fig. 2D; n = 4; P > 0.05, +50.4 ± 51.2, Pro versus Ala; Seabold et al., 2003). Altogether, these data indicate that Pro is specific for its capability to interfere with the ADAM10/SAP97 complex.
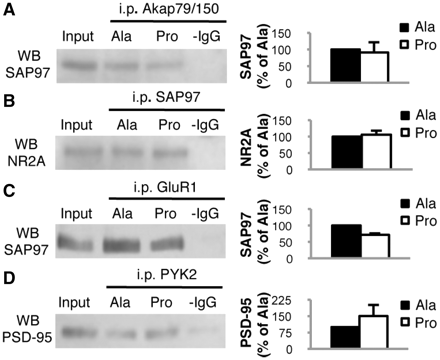
Pro peptide treatment does not interfere with protein–protein complexes other than ADAM10/SAP97. (A) C57BL/6 mice were treated for 2 weeks with Pro or Ala peptides and total brain homogenates were co-immunoprecipitated with anti-Akap79/150 antibody. SAP97 presence was evaluated in the immunocomplex. The association is not altered by treatment. (B) C57BL/6 mice were treated for 2 weeks with Pro or Ala peptides and total brain homogenates were co-immunoprecipitated with anti-SAP97 antibody. NR2A presence was evaluated in the immunocomplex. The association is not altered by treatment. (C) C57BL/6 mice were treated for 2 weeks with Pro or Ala peptides and total brain homogenates were co-immunoprecipitated with anti-GluR1 antibody. SAP97 presence was evaluated in the immunocomplex. The association is not altered by treatment. (D) C57BL/6 mice were treated for 2 weeks with Pro or Ala peptides and total brain homogenates were co-immunoprecipitated with anti-PYK2 antibody. PSD-95 presence was evaluated in the immunocomplex. The association is not altered by treatment. For all experiments, quantitative analysis of immunostaining is shown as percentage of Ala treatment in the same experiment. i.p. = immunoprecipitation; WB = western blot.
Treatment with Pro alters ADAM10 localization and generates the characteristic features of Alzheimer's disease: amyloidogenesis and tau hyperphosphorylation
SAP97 is necessary to drive ADAM10 to the plasma membrane (Marcello et al., 2007). We asked whether the reduction in ADAM10/SAP97 association, obtained through Pro peptide treatment, could induce a modification in ADAM10 localization. Western blot analysis performed on TIF—a fraction enriched in proteins of the postsynaptic density (Gardoni et al., 2001)—revealed a statistically significant reduction of ADAM10 levels in mice treated with Pro compared with mice treated with Ala (Fig. 3A; n = 4; P = 0.006, −16.7 ± 2.2%, Pro versus Ala). No changes were observed in SAP97 immunoreactivity in the TIF (Fig. 3A; n = 4, P > 0.05, −10.4 ± 5.1%, Pro versus Ala).
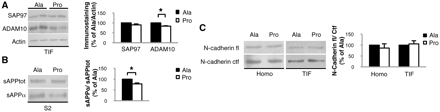
Pro treatment alters ADAM10 localization and activity on amyloid precursor protein. (A) Western blot of TIF from Pro- and Ala-treated mice performed with anti-ADAM10 and anti-SAP97 antibodies. Pro treatment reduced ADAM10 levels (P = 0.006) without modifying SAP97 levels. All data were normalized to actin. (B) Western blot performed on the soluble fraction S2 of mice treated with Pro or Ala peptides. sAPPα/sAPPtot ratio is reduced in mice treated with Pro peptide (P = 0.043). (C) Western blot performed on total brain homogenate and TIF fraction of Pro- and Ala-treated mice to reveal N-cadherin full-length (fl) and C-terminal fragment (Ctf). N-cadherin fl/Ctf ratio was not altered by the treatment with Pro peptide. For all experiments, quantitative analysis of immunostaining is shown as percentage of Ala treatment in the same experiment.
Since ADAM10 is active at the plasma membrane (Lammich et al., 1999), we supposed that the reduction of ADAM10 in the postsynaptic compartment could be paralleled by a decrease in its activity towards its synaptic substrates. sAPPα release, deriving from ADAM10 cleavage on amyloid precursor protein, was decreased in Pro-treated compared with Ala-treated mice (Fig. 3B; n = 4, P = 0.043, −22.7 ± 4.6%, Pro versus Ala), whereas we observed no alterations in the processing of N-cadherin, another ADAM10 synaptic substrate (Reiss et al., 2005; Fig. 3C; n = 4; homogenate: −9.1 ± 19.6%; TIF: +5.7 ± 10.5%; overall P > 0.05, Pro versus Ala).
To exclude compensatory upregulation of other metalloproteases after 2 weeks of treatment, levels of matrix metallopeptidase 9 (MMP-9) and its gelatinolytic activity were analysed. No alterations were found in MMP-9 total brain homogenate levels or in its gelatinolytic activity (Supplementary Fig. 2A; n = 4; homogenate: +12.2 ± 5.6%, P > 0.05; Supplementary Fig. 2B; n = 4; zymography: −8.7 ± 6.5%, P > 0.05; Pro versus Ala).
Altered amyloid precursor protein metabolism induced by Pro peptide treatment was further investigated. We measured amyloid precursor protein CTFs: CTF83 for α-secretase and CTF99 for β-secretase cleavage, in TIF. The CTF83/CTF99 ratio was reduced in mice treated with Pro compared with mice treated with Ala, indicating a shift of amyloid precursor protein metabolism towards amyloidogenesis in Pro-treated mice (Fig. 4A; n = 4, P = 0.03, −28.6 ± 5.7%, Pro versus Ala). The shift of amyloid precursor protein metabolism towards Aβ production was further confirmed by the raise in Aβ1–42 production detected by ELISA in total brain homogenate of mice treated with Pro if compared with Ala-treated mice (Fig. 4B, n = 4; P = 0.02; +16.8 ± 4.6%, Pro versus Ala).
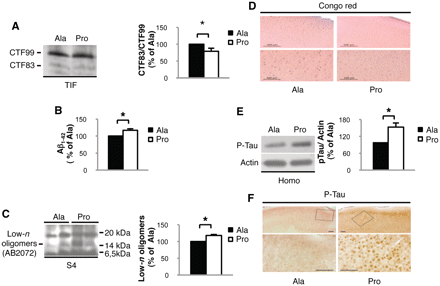
Pro treatment induces Alzheimer's pathological hallmarks. (A) Western blot performed on the TIF of Pro- and Ala-treated mice to reveal amyloid precursor protein CTFs. CTF83/CTF99 ratio is decreased in mice treated with Pro peptide (P = 0.03). (B) Quantification of Aβ1-42 production in mice treated with Pro compared with Ala. Aβ1-42 production is increased after Pro treatment (P = 0.02). (C) Western blot performed on the formic acid soluble fraction S4 of Pro- and Ala-treated mice to reveal Aβ oligomers. A 16 kDa band (at theoretical apparent molecular mass of tetramers) is increased after Pro treatment (P = 0.0005). (D) Representative images of Congo Red staining showing no senile plaques in Pro- and Ala-treated mice (scale bar = 500 µm and 200 µm). (E) Western blot performed on homogenate from mice treated with Pro or Ala peptides. Tau phosphorylation is increased in Pro-treated mice (P = 0.038). Data were normalized to actin. (F) Representative images of phosphorylated-tau (P-Tau) staining showing increased phosphorylation in Pro-treated mice (scale bar = 100 µm). For all experiments, quantitative analysis of immunostaining is shown as percentage of Ala treatment in the same experiment.
An initiating factor in Alzheimer's disease pathogenesis occurs when soluble, monomeric Aβ undergoes a conformational change and converts into oligomeric forms (Klein et al., 2001). Although murine Aβ is less prone to aggregation than human Aβ, we detected low-n Aβ oligomers in formic acid soluble extracts obtained by sequential centrifugation of brain homogenates (Lesne et al., 2006). An antibody raised against the N-terminus of Aβ, AB2072, revealed an increase of an oligomeric aggregate at 16 kDa, a molecular mass corresponding to tetramers (Fig. 4C; n = 4, P = 0.0005, +18.3 ± 1.8%, Pro versus Ala) in Pro-treated mice. The detection of this band with AB2072 and 4G8, recognizing Aβ mid-domain, excludes the possibility that it represents degradation products of soluble amyloid precursor protein. The band was not recognized by antibodies for epitopes at the C-terminus or the N-terminus of amyloid precursor protein, amyloid precursor protein C-Term and 22C11, respectively, indicating it was neither one of the amyloid precursor protein CTFs nor amyloid precursor protein cleavage-end products (Supplementary Fig. 3A and Supplementary Methods). As revealed by Congo Red staining, we detected no plaques in Pro- and Ala-treated mice (Fig. 4D), suggesting that we are mimicking the early stages of Alzheimer's disease (Oddo et al., 2003).
Since Aβ production triggers the development of other Alzheimer's disease hallmarks, such as neurofibrillary tangles (Oddo et al., 2003), we evaluated the presence of tau hyperphosphorylation. Tau phosphorylation in serine202–205 was increased in mice treated with Pro peptide (Fig. 4E; n = 4, P = 0.038, +53.2 ± 15.0%, Pro versus Ala). This was corroborated by immunohistochemical analysis showing tau hyperphosphorylation in soma, axons and dendrites of neurons in the cerebral cortex of Pro-treated mice compared with Ala-treated ones (Fig. 4F). Tau-C3 antibody is specific for tau cleaved at aspartic acid421, which represents a further stage of neurofibrillary tangle evolution in Alzheimer's disease (Gamblin et al., 2003). Tau-C3 staining revealed no signal in both groups (Supplementary Fig. 3B).
Effects of Pro peptide treatment on spine morphology and on molecular composition of the excitatory glutamatergic synapse
Synaptic dysfunction appears as an early event in disease pathogenesis with synapse loss evident in patients at early stages of Alzheimer's disease and with mild cognitive impairment (Scheff et al., 2007). We evaluated potential alterations of synaptic morphology associated with the observed shift of amyloid precursor protein metabolism towards amyloidogenesis. Analysis of dendritic spines was performed using DyOlistic fluorescent labelling (Gan et al., 2000). We measured density (N/10 µm), area, neck length and head width of dendritic spines in the hippocampus and cortex. Statistical analysis showed no differences in all parameters between animals treated with Pro and Ala peptides, indicating that we are analysing the early stages preceding spine loss and degeneration [Fig. 5A; (i) cortex Ala: n = 33, area 0.384 ± 0.009 µm2, length 1.29 ± 0.02 µm, width 0.53 ± 0.01 µm, spines/10 µm 10.67 ± 0.42; Pro: n = 33, area 0.348 ± 0.01 µm2, length 1.30 ± 0.01 µm, width 0.557 ± 0.01 µm, spines/10 µm 10.53 ± 0.44; overall P > 0.05; Fig. 5B; (ii) hippocampus Ala: n = 33; area 0.312 ± 0.009 µm2, length 0.884 ± 0.01 µm, width 0.483 ± 0.008 µm, spines/10 µm 12.31 ± 0.573; Pro: n = 33, area 0.307 ± 0.008 µm2, length 0.84 ± 0.01 µm, width 0.474 ± 0.007 µm, spines/10 µm 12.10 ± 0.48; overall P > 0.05].
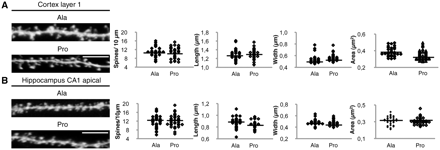
Effects of Pro treatment on spine density and morphology. Representative images of cortical layer I (A) and hippocampal CA1 apical (B) spines from Pro- and Ala-treated mice. Statistical analysis of spine density, neck length, head width and area revealed no modifications in both areas (scale bar = 5 µm).
To characterize molecular events occurring prior to synaptic loss, we evaluated biochemically the composition of glutamatergic postsynaptic density. No alterations in PSD-95 (Fig. 6A; n = 4; homogenate: +12.7 ± 6.1%; TIF: +1.2 ± 6.6%; overall P > 0.05, Pro versus Ala) and α-CaMKII (Fig. 6A; n = 4; homogenate: −1.1 ± 2.7%; TIF: +30.2 ± 18.9%; overall P > 0.05, Pro versus Ala) levels were found, confirming no major modifications of postsynaptic density organization in early stages of Aβ formation. We detected no significant alterations in GluR1 (Fig. 6A; n = 4; homogenate: +18.6 ± 26.0%; TIF: +20.3 ± 23.1%; overall P > 0.05, Pro versus Ala) and GluR2 (Fig. 6A; n = 4; homogenate: +0.9 ± 10.9%; TIF: +25.3 ± 7.2%; overall P > 0.05, Pro versus Ala) AMPA receptor subunit levels in total homogenate and TIF in Pro-treated animals compared with Ala-treated ones. NR1, the NMDA receptor constitutive subunit, was unaltered (Fig. 6B; n = 4; homogenate: +19.4 ± 14.7%; TIF: +20.2 ± 8.0%; overall P > 0.05, Pro versus Ala) as was NR2B (Fig. 6B; n = 4; homogenate: −2.6 ± 5.4%; TIF: +7.4 ± 7.8%; overall P > 0.05, Pro versus Ala), but a specific reduction of NR2A subunit in TIF was observed (Fig. 6B; n = 4; homogenate: +17.1 ± 8.4%, P > 0.05; TIF: −27.4 ± 2.8%, P = 0.002, Pro versus Ala).]
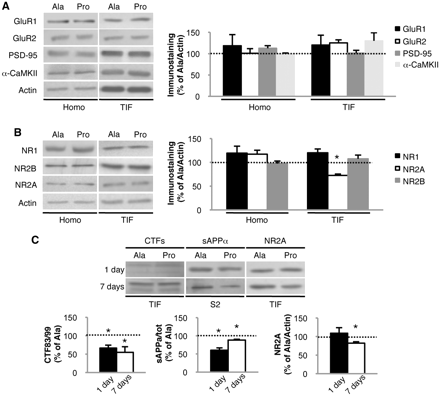
Effects of Pro treatment on the glutamatergic synapse. (A) Western blot of homogenate (Homo) and TIF from Pro- and Ala-treated mice performed with anti-GluR1, anti-GluR2, anti-PSD-95 and anti-α-CaMKII antibodies. (B) Western blot of homogenate and TIF from Pro- and Ala-treated mice performed with anti-NR1, anti-NR2A and anti-NR2B antibodies. NR2A is decreased in the TIF (P = 0.002). (C) Western blot analysis of the TIF and soluble fraction S2 of mice treated for 1 or 7 days with Pro or Ala. The CTF83/CTF99 ratio is reduced in mice treated with Pro peptide for 1 day (P = 0.01) and 7 days (P = 0.035). sAPPα/sAPPtot ratio is reduced in mice treated with Pro peptide for 1 day (P = 0.02) and 7 days (P = 0.015). NR2A is reduced in the TIF after 7 days of treatment with Pro (P = 0.01) but not after 1 day. All data were normalized to actin. For all experiments, quantitative analysis of immunostaining is shown as percentage of Ala treatment in the same experiment.
This modification of the molecular composition of glutamatergic postsynaptic density in Pro-treated mice temporally follows Aβ production. We evaluated the ADAM10/SAP97 immunocomplex (1 day: P = 0.008, −32.4 ± 3.9%; 7 days: P = 0.003, −25.4 ± 2.4%, Pro versus Ala), ADAM10 in TIF (1 day: P = 0.04, −21.3 ± 2.7%; 7 days: P = 0.019, −17.5 ± 4.2%, Pro versus Ala) as well as amyloid precursor protein metabolites at Days 1 and 7 of treatment. sAPPα/total sAPP and CTF83/CTF99 ratios were shifted towards amyloidogenesis after both 1 and 7 days of treatment with the Pro peptide (Fig. 6C; 1 day: n = 4; sAPPalpha/tot: P = 0.02, −39.7 ± 5.2%; CTF83/99: P = 0.01, −34.3 ± 7.9%, Pro versus Ala; 7 days: n = 4; sAPPalpha/tot: P = 0.015, −10.1 ± 2.3%; CTF83/99: P = 0.035, −45.8 ± 15.2%, Pro versus Ala). In parallel, NR2A levels in TIF were measured. After 7 days, but not 1 day, of Pro peptide treatment, NR2A decreased in postsynaptic density (Fig. 6C; n = 4, 1 day: P > 0.05, +9.1 ± 14.8%; 7 days: P = 0.01, −17.8 ± 3.5%, Pro versus Ala).
These results suggest that increases in Aβ production precede alterations of excitatory synapses.
Functional deficits: electrophysiological and behavioural consequences of Pro peptide treatment
We then evaluated activity-dependent synaptic plasticity. We tested long-term potentiation in the CA1 region of acute hippocampal slices obtained from mice treated with either the Pro or Ala peptide for 14 days. When potentiation of CA3–CA1 synapses was induced by high-frequency stimulation, no effects of peptide treatment were found on the magnitude of long-term potentiation measured 50–60 min after high-frequency stimulation. However, the maximum potentiation of the synaptic response immediately following high-frequency stimulation was significantly reduced in Pro-treated mice (Fig. 7A; P < 0.02, ANOVA, Pro: 278.8 ± 37.89%; Ala: 418.70 ± 29.27%). This reduction of synaptic plasticity induced by the Pro peptide was statistically significant up to 10 min after high-frequency stimulation, a period in which enhancements of synaptic responses can be attributable to the buildup and decay of post-tetanic potentiation (Zucker and Regehr, 2002), a form of short-term synaptic plasticity.
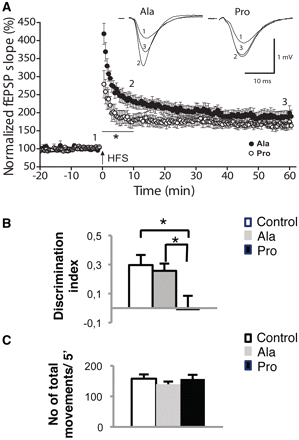
Effects of Pro-treatment on glutamatergic synapse function. (A) A reduction in post-tetanic potentiation induced by high-frequency stimulation (HFS; 4 × 100 Hz, 1 s, 30 s apart) was observed in Pro-treated mice (open circles, n = 8 slices from n = 3 mice) compared with Ala-treated (full circles, n = 8 slices from n = 3 mice; P < 0.01, ANOVA). Long-term potentiation measured 50–60 min after HFS was similar in the presence of either peptide. Insets illustrating averaged extracellular field excitatory postsynaptic potentials from three consecutive traces taken before (1) and at different times after HFS (2 and 3) are shown above summary data. Error bars indicate SEM. (B) Quantification of the discrimination index of mice treated with Pro or Ala peptides in the novel object recognition test. Mice treated with Pro peptide show a reduced discrimination index compared with those treated with Ala peptide (P = 0.042, ANOVA) and with saline-treated mice (P = 0.021, ANOVA). (C) Quantification of the number of movements in 5 min in mice treated with Pro or Ala peptides in the Novel Object Recognition test. All behavioural data are expressed as means ± SEM (n = 9 per group).
To assess behavioural dysfunctions paralleling the alterations in synaptic organization and function, we performed a Novel Object Recognition test on animals treated with Pro or Ala for 2 weeks. Treated mice were unable to distinguish new objects from known ones, with no significant difference in the percentage of time spent investigating the two objects. Discrimination index between novel and familiar objects revealed a prominent effect of treatment with the Pro peptide. Post hoc analysis found a significant inability to discriminate between the two objects in Pro-treated mice [Fig.7B; discrimination index, Ala: 0.26; Pro: −0.01; saline: 0.3; ANOVA (Pro/Ala) P = 0.04]. To assess whether administration of the peptides influenced the mobility of mice, the number of total movements in 5 min was determined and no significant differences were found between the groups [Fig. 7C; number of movements/5 min, Ala: 139.8 ± 8.5; Pro: 156.6 ± 13.7; saline: 157.8 ± 14.0; ANOVA (Pro/Ala) P > 0.05].
Discussion
Development of adequate animal models mimicking all stages of Alzheimer's disease progression and merging convergent pathways of pathogenesis still represents a need for research on Alzheimer's disease. Available mouse models have been generated on the basis of identified human mutations (Goate et al., 1991; Scheuner et al., 1996). Although informative for the understanding of molecular pathogenesis of the disease, these models present a number of weaknesses (Epis et al., 2010; Morrissette et al., 2009). In transgenic models, mutated proteins are chronically overexpressed and exposure to Aβ overproduction starts before the common clinical onset of Alzheimer's disease. The genetic strains, the mutations and the different transcriptional promoters used to drive their expression, as well as other factors that are not completely known, produce different amounts of Aβ, forming potentially different oligomeric forms, which may induce high variability and different outcomes across transgenic mouse models (Epis et al., 2010; Gerlai, 1996; Morrissette et al., 2009).
Here, we propose a different approach to investigating the consequences of Aβ increase in an innovative, non-transgenic animal model of early Alzheimer's disease.
Our strategy is based on interference with the ADAM10/SAP97 protein complex in adult mice. We have already reported the interaction with the PSD-MAGUK protein SAP97 as pivotal for ADAM10 trafficking to the postsynaptic membrane and for its enzymatic non-amyloidogenic cleavage on amyloid precursor protein (Marcello et al., 2007). The relevance of the ADAM10/SAP97 complex in early stages of disease is strengthened by our analysis of the hippocampus of patients at Braak level 4 of Alzheimer's disease, where we observed a clear reduction in ADAM10/SAP97 co-immunoprecipitation. These results suggest that the defect of interaction between the two proteins is relevant for human pathology.
Through administration of a cell-permeable peptide, Pro, for 2 weeks, we were able to reproduce ADAM10/SAP97 uncoupling in vivo in rodent brains. In Pro peptide, the short Tat sequence (11 amino acids) (Aarts et al., 2002) is fused to the 32 amino acid sequence of the proline-rich domains of ADAM10, responsible for the direct binding to the SH3 domain of SAP97 (Marcello et al., 2007). The peptide does not interfere with other protein complexes, implying that reported functional and behavioural modifications are not related to off-target effects.
Treatment of mice for 2 weeks with Pro peptide reduces ADAM10/SAP97 association and, consequently, ADAM10 localization to the postsynaptic membrane.
Specificity of the effects obtained after peptide treatment remains high with regard to the large number of ADAM10 substrates already described in the literature. This could be ascribed to either: (i) not all ADAM10 substrates are neuronally expressed; or (ii) our model is fully based on the uncoupling of the ADAM10/SAP97 complex in a very confined structure, the glutamatergic synapse, where both proteins are located. Again, among neuronal ADAM10 substrates, not all have spine localization.
Pro peptide treatment influences amyloid precursor protein metabolism, reducing α-secretase-mediated processing without affecting the processing of other synaptic, but developmentally relevant substrates, such as N-cadherin. The molecular rationale supporting the lack of ADAM10 cleavage of N-cadherin in the mature spine is at present not completely clear. Nonetheless, we can speculate that either clusterization of the enzyme or different accessibility of the substrates in mature spines compared with developmental stages could be relevant.
Importantly, there are no specific chemical inhibitors for basal ADAM10/α-secretase activity (Ludwig et al., 2005). Pro peptide could therefore represent an appropriate tool to study the physiological role of ADAM10.
Exposing mice to Pro peptide for 2 weeks affects amyloid precursor protein metabolism, increasing amyloid precursor protein amyloidogenic cleavage with a modest but significant raise of Aβ release and of low-n Aβ aggregate production. This subtle increase in Aβ appears particularly promising when compared with data collected from studies in engineered animal models, where accumulation of soluble Aβ is normally reported in older animals, i.e. 9–13 months of age (Holcomb et al., 1998; Oddo et al., 2003a). Similarly, even in triple transgenic mice, Aβ oligomers can be revealed after 13 months of age (Oddo et al., 2003,b, 2006). Notably, in our conditions, the slight increase of both soluble Aβ and its low-n aggregates is evident after just 2 weeks of treatment. Although the low aggregation capability of rodent Aβ can be considered a limitation, ADAM10/SAP97 uncoupled animals still appear as a promising model to study early phases of Alzheimer's disease.
From a pathological point of view, hallmarks of Alzheimer's disease include extracellular amyloid plaques as well as intracellular neurofibrillary tangles (Selkoe, 2001). The latter trait is characteristic of human Alzheimer's disease but is not always present in amyloid models (Morrissette et al., 2009), which in most cases do not even show tau hyperphosphorylation. For instance, mutant presenilin-1 mice crossed with mutant amyloid precursor protein mice fail to develop detectable neurofibrillary tangles (Holcomb et al., 1998) and also in triple transgenic mice, lesions accumulate in an age-dependent fashion (Oddo et al., 2003a). In ADAM10/SAP97 uncoupled mice, tau hyperphosphorylation, but not neurofibrillary tangles, was already evident after 2 weeks of treatment with Pro peptide, further supporting our hypothesis that these mice mimic early phases of pathology.
However, rather than deposition of amyloid plaques or neurofibrillary tangles, the most robust correlation in the staging of dementia and early Alzheimer's disease is the magnitude of synapse loss (Terry et al., 1991; Scheff et al., 2007). Spine loss and alterations in morphology have been linked to Aβ and its oligomeric aggregates in in vivo and in vitro studies (Lacor et al., 2004; Shankar et al., 2008; Wei et al.), suggesting a convergence between the main causes of Alzheimer's disease pathogenesis: amyloid cascade and synaptic alterations. In ADAM10/SAP97 uncoupled mice, we failed to adversely affect spine number and to alter spine morphology, probably because of the limited time of in vivo exposure to Aβ and its aggregates. The reasons for spine alterations in Alzheimer's disease remain controversial. Data emerging from a variety of transgenic mouse lines have pointed out a role for both low- and high-number oligomers (Jacobsen et al., 2006). Their mechanism of action remains elusive, mostly because the vast majority of studies on the effects of Aβ oligomers on spine morphology have been carried out in vitro using in vivo-produced oligomers. A better knowledge of the mechanisms underlying spine deterioration in patients with Alzheimer's disease, which might involve effects of Aβ on cytoskeletal (Alvarez and Sabatini, 2007) and mitochondrial dysfunction (Hauptmann et al., 2009), would be crucial for speculations on the reasons for the lack of spine alterations in our model. Studies with longer exposure to the Pro peptide might be helpful to dissect structural effects of ADAM10/SAP97 uncoupling.
Nevertheless, in ADAM10/SAP97 uncoupled mice, synaptic sufferance occurs prior to anatomical changes and is evident as a reorganization of the molecular composition of excitatory glutamatergic synapse, with a selective reduction of the NR2A NMDA subunit. Recently, an effect of Aβ oligomers on NMDA receptor subunits has been reported, revealing an effect on NR2B trafficking and internalization (Snyder et al., 2005; Lacor et al., 2007). Our current findings on NR2A and spine morphology seem apparently contradictory, when compared with recent literature. This could be due to experimental discrepancies among studies, including different Aβ species, different stages of pathology or mice development at exposure and different concentrations and/or oligomerization state of Aβ (Billings et al., 2005). The site of action of Aβ should also be considered, since intracellular and extracellular pools of Aβ may differently influence synaptic function, and the net outcome may depend on their dynamic relationship (Venkitaramani et al., 2007). However, in agreement with previous studies (Cleary et al., 2005), in our experimental conditions, NMDA subunit modifications at synaptic sites follow Aβ formation and accumulation, suggesting Aβ formation as the starting event of pathogenesis.
In our model, we described activity-dependent alterations of synaptic plasticity as well as an initial cognitive deficit in the Novel Object Recognition test, paralleling NR2A reduction. Interestingly, recent reports showed that long-term potentiation impairment in transgenic mice models of Alzheimer's disease depends on the age of the animals and, presumably, on Alzheimer's disease stage. For example, studies in ArcAb mice (where a form of Aβ more prone to yielding Aβ oligomers is expressed) revealed that long-term potentiation is impaired in mice of 3.5 and 7.5 months of age, but not in those of 1 month, probably because at this age, Aβ accumulation is not yet detectable (Knobloch et al., 2007). In light of these previous data, lack of differences in hippocampal long-term potentiation in our model could be correlated to the early stage of Alzheimer's disease pathology revealed by our biochemical and morphological results. On the other hand, alterations in short-term synaptic plasticity found in our model have been documented in other Alzheimer's disease models. For example, double transgenic mice overexpressing the mutated form of amyloid precursor protein (APPK670N, M671L) and presenilin-1 (PS1M146V) exhibit enhanced synaptic fatigue, a form of short-term synaptic plasticity, at the Schaffer collateral-CA1 synapse (Zhang et al., 2005). Intriguingly, because short-term synaptic changes are relevant in cognitive processes such as learning and memory, it is conceivable that alterations in the synaptic plasticity shown by our Alzheimer's disease model could be one leading cause of the impaired behavioural performances of these animals. Indeed, Pro peptide treated mice showed a marked impairment in the Novel Object Recognition test, which measures recognition memory and is heavily altered in Alzheimer's disease. This test relies on spontaneous animal behaviour without the need for stressor elements. Pro peptide treatment impairs memory consolidation within 24 h and rapidly interferes with the synaptic activity necessary for the stabilization of new memories.
In conclusion, our model can be used to study pathogenic consequences of a specific pathway of Alzheimer's disease pathogenesis, namely ADAM10/SAP97 disruption. Pro peptide treated animals manifest all characteristics of the early stages of Alzheimer's disease, i.e. increased Aβ production and presence of low-n aggregates, hyperphosphorylated tau and clear synaptic dysfunction as reflected by aberrant composition of NMDA receptors, altered activity-dependent plasticity and impaired behavioural performance. These manifestations occur prior to synapse loss and spine modifications and before the appearance of full pathological hallmarks, i.e. amyloid plaques and neurofibrillary tangles (Selkoe, 2008), highlighting the relevance of our model in studying the early stages of Alzheimer's disease.
Pro peptide strategy offers the unprecedented possibility to interfere with endogenous amyloid precursor protein processing in adult mice by neither overexpressing proteins derived from inherited forms of the disease nor altering developmental processes. Moreover, it can be useful to study the convergence as well as the temporal correlation of multiple neuropathological pathways, such as Aβ production and synaptic dysfunction and, eventually, to investigate new therapeutic strategies.
Funding
The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7 2007-2013; Grant Agreement n° PIAP-GA-2008-217902 to M.D.L.); by Fondazione Cariplo 2008 to M.D.L.; by Telethon-Italy (Grant Number GGP09196 to M.G.); by Compagnia di San Paolo to M.G. and by a fellowship from the Spanish Ministerio de Educación y Ciencia (Grant Number EX2006-0294 to H.V.). The authors are grateful to E. Zianni and A. Longhi for skilful technical work.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- ADAM10
a disintegrin and metalloproteinase domain with protein 10
- Aβ
β-amyloid peptide
- Ala
Tat-Ala ADAM10709-729 peptide
- CTF
carboxy terminal fragment
- Pro
Tat-Pro ADAM10709-729 peptide
- SAP97
synapse-associated protein 97
- TIF
Triton-X insoluble fraction
References
Author notes
*These authors contributed equally to this work.
†Present address: Instituto de Neurociencias de Alicante, Universidad Miguel Hernández – CSIC. Sant Joan d'Alacant, 03550 Alicante, Spain.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}