




Abstract
In a genome-wide association study of frontotemporal lobar degeneration with pathological inclusions of TAR DNA-binding protein, significant association was obtained with three single nucleotide polymorphisms at 7p21.3, in a region encompassing the gene TMEM106B. This study also suggested a potential modifying effect of TMEM106B on disease since the association was strongest in progranulin mutation carriers. Further, the risk effect seemed to correlate with increased TMEM106B expression in patients. In the present study, we sought to replicate these three findings using an independent Flanders–Belgian cohort of primarily clinically diagnosed patients with frontotemporal lobar degeneration (n = 288). We were able to confirm the association with TMEM106B with a P-value of 0.008 for rs1990622, the top marker from the genome-wide association study [odds ratio 0.75 (95% confidence interval 0.61–0.93)]. Further, high-density single nucleotide polymorphism mapping suggested that the association was solely driven by the gene TMEM106B. Homozygous carriers of the TMEM106B protective alleles had a 50% reduced risk of developing frontotemporal lobar degeneration. However, we were unable to detect a modifying effect of the TMEM106B single nucleotide polymorphisms on onset age in progranulin mutation carriers belonging to an extended, clinical and pathological well-documented founder family segregating a progranulin null mutation. Also, we could not observe significant differences in messenger RNA expression between patients and control individuals in lymphoblast cell lines and in brain frontal cortex. In conclusion, we replicated the genetic TMEM106B association in a primarily clinically diagnosed cohort of patients with frontotemporal lobar degeneration from Flanders–Belgium. Additional studies are needed to unravel the molecular role of TMEM106B in disease onset and pathogenesis.
Introduction
Since frontotemporal lobar degeneration (FTLD) was linked to chromosome 17, enormous progress has been made in the molecular genetics and neuropathology of FTLD providing valuable insights into its disease pathogenesis. However, yet the underlying disease mechanisms of FTLD are not fully understood and no effective treatments are available. Two major pathological subtypes exist i.e. FTLD with cellular inclusion bodies of tau (FTLD-tau) or TDP-43 (FTLD-TDP), each representing 40–50% of patients (Mackenzie et al., 2010). Whereas pathogenic mutations in the gene coding for the microtubule associated protein tau (MAPT) is associated with FTLD-tau, FTLD-TDP pathology is observed in patients carrying a mutation in progranulin (GRN) or in the valosin-containing protein gene (VCP) (Mackenzie et al., 2010).
An international collaborative genome-wide association study, aimed at identifying genetic risk factors influencing FTLD-TDP pathology, was performed in patients with either autopsy confirmed FTLD-TDP or a clinical diagnosis of FTLD in the presence of a GRN mutation (Van Deerlin et al., 2010). Three single nucleotide polymorphisms (SNPs) reached genome-wide significance (rs1020004, rs6966915 and rs1990622) and mapped to a single linkage disequilibrium block of 68 kb at 7p21.3. The genomic region encompassed the gene coding for TMEM106B, an uncharacterized transmembrane protein, and two other genes at the rim of the linked region, scinderin (SCIN) and ADP-ribosylation factor-like 4A (ARL4A). The effect appeared to be most pronounced in carriers of pathogenic GRN mutations, suggesting a modifying effect of TMEM106B in GRN mutation carriers. Furthermore, messenger RNA quantifications showed >2.5-fold increased TMEM106B expression in frontal cortex of patients with FTLD-TDP compared with unaffected individuals. These findings suggested that the increased TMEM106B expression in brain could be linked to FTLD-TDP pathogenesis, however, they were based on a modest number of brain samples (n = 25).
It is commonly accepted that newly identified risk genes, such as those identified in genome-wide association studies of complex diseases, can only be established if they are replicated in independent study populations. Therefore, we set out to replicate the FTLD association with the TMEM106B locus in a Flanders–Belgian cohort of patients and control individuals. In contrast to the original study, which selected patients with FTLD-TDP pathology, we used a selection of patients with primarily clinical diagnoses of FTLD.
Materials and methods
Study cohorts
Patients with FTLD included in this study were recruited from 1998 onwards in Flanders–Belgium as part of an ongoing collaboration of neurology departments and memory clinics of the Hospital Network Antwerp (ZNA) Middelheim and ZNA Hoge Beuken, the University Hospital Antwerp, the University Hospitals Leuven and the University Hospital Ghent. All index patients were evaluated using a standard diagnostic protocol including neurological examination, neuropsychological testing, biochemical analyses, EEG and neuroimaging. Clinical diagnosis was made according the Neary consensus criteria for FTLD (Neary et al., 1998, 2005) and patients were followed longitudinally on a regular basis. Plasma, serum and cerebrospinal fluid samples were collected. In a subset of patients, neuropathological examination of autopsied brain was performed to obtain diagnostic certainty and to define the FTLD pathology subtype (Engelborghs et al., 2008).
Index patients collected in the clinics as well as patients who were referred for genetic testing to our diagnostic service facility, were contacted to obtain consented participation in molecular genetic studies of neurodegenerative dementias. Blood samples from patients, their relatives and from control individuals were collected for DNA extraction, establishing Epstein-Barr virus transformed lymphoblast cell lines and the collection of serum and plasma. Neurologically healthy community control individuals were ascertained in Flanders–Belgium and underwent a medical interview questioning their personal, medical and family history of neurological or psychiatric disease followed by a mini-mental state examination. Brain autopsy and pathological testing was proposed to patients and/or their legal guardians as well as to control individuals.
All participants gave written informed consent for participation in both the clinical and genetic studies. The clinical study protocol and the informed consent forms for patient ascertainment were approved by the Medical Ethic Committees of ZNA Middelheim and ZNA Hoge Beuken, University Hospital Antwerp, University Hospitals Leuven and University Hospital Ghent. The genetic study protocol and informed consent forms were approved by the Medical Ethical Committee, University of Antwerp, Belgium. Patient relatives and control individuals were ascertained after written informed consent, within the frame of the medical ethical approved genetic study protocol. All samples received a unique identifier number and demographic, medical and genetic data were stored in a centralized database. Restrictive access permissions were given to researchers and clinicians depending on their respective role in the study.
Study subjects
From the Flanders–Belgian FTLD study population of 315 patients, 297 unrelated patients with a clinical diagnosis of FTLD were selected for the purpose of this study (Table 1). All patients were analysed for mutations in the four FTLD genes—MAPT, GRN, VCP and CHMP2B (Hutton et al., 1998; Watts et al., 2004; Skibinski et al., 2005; Baker et al., 2006; Cruts et al., 2006). We excluded patients with known non-TDP pathology, patients carrying a mutation in MAPT or CHMP2B (van der Zee et al., 2008) or having an autopsy diagnosis other than FTLD-TDP. The final FTLD patient cohort counted 13 unrelated patients carrying a GRN loss-of-function mutation including one patient of the GRN IVS1 + 5G > C founder family (Cruts et al., 2006; Gijselinck et al., 2008), and two unrelated patients with the VCP R159H mutation (van der Zee et al., 2009). Twenty-two patients had a clinical diagnosis of FTLD with amyotrophic lateral sclerosis, and 14 patients had a pathology diagnosis of FTLD-TDP. Average onset age was 62.8 ± 10.7 years and ranged from 29 to 90 years. At least one other affected relative was reported in 33.7% of patient’s medical records. Nine of the 297 patients were part of the original FTLD-TDP genome-wide association study (Van Deerlin et al., 2010) and were excluded from the replication analysis. After we confirmed association of TMEM106B with FTLD in our Flanders–Belgian cohort, we included all 297 patients in the SNP fine-mapping. To maximize the power to find disease association, we used a control population twice the size of the patient sample. The 595 age-matched control individuals had an average age at inclusion of 61.8 ± 15.4 years ranging from 19–94 years (Table 1). Only disease-free individuals without family history of neurodegenerative brain diseases and a mini-mental state examination score > 24 were included in the control sample. Together this patient–control cohort offered > 99% power to detect disease association of a variant with minor allele frequency of 0.4 and odds ratio of 0.6 [values based on the findings of Van Deerlin et al. (2010), calculated with the Genetic Power Calculator (Purcell et al., 2003)].
Description of the Flanders–Belgian FTLD patient and control cohort
| Parameters | Flanders-Belgian FTLD cohort | FTLD patient selection for current study | Flanders–Belgian control cohort | Control individual selection for current study |
|---|---|---|---|---|
| N | 315 | 297c | 870 | 595 |
| Age at onset/inclusiona | 62.3 ± 10.6 (29–90) | 62.8 ± 10.7 (29–90) | 65.2 ± 14.9 (19–94) | 61.8 ± 15.4 (19–94) |
| Familialb, n (%) | 119 (37.7%) | 100 (33.7) | NA | NA |
| Mutations, n (%) | 30 (9.5) | 15 (5.1) | NA | NA |
| Pathology diagnoses, n (%) | 22 (7.0) | 14 (4.7) | NA | NA |
| Parameters | Flanders-Belgian FTLD cohort | FTLD patient selection for current study | Flanders–Belgian control cohort | Control individual selection for current study |
|---|---|---|---|---|
| N | 315 | 297c | 870 | 595 |
| Age at onset/inclusiona | 62.3 ± 10.6 (29–90) | 62.8 ± 10.7 (29–90) | 65.2 ± 14.9 (19–94) | 61.8 ± 15.4 (19–94) |
| Familialb, n (%) | 119 (37.7%) | 100 (33.7) | NA | NA |
| Mutations, n (%) | 30 (9.5) | 15 (5.1) | NA | NA |
| Pathology diagnoses, n (%) | 22 (7.0) | 14 (4.7) | NA | NA |
Mutations in the Flanders–Belgian FTLD cohort: 22 GRN (7.0%), 4 MAPT (1.3%), 2 VCP (0.6%), 1 CHMP2B (0.3%) and 1 PSEN1 (0.3%).
Pathology diagnoses in the Flanders–Belgian FTLD cohort: 19 FTLD-TDP (6.0%), 1 FTLD-tau (0.3%), 1 FTLD-UPS (0.3%), 1 FTLD-ni (0.3%).
a Age at onset for patients or age at inclusion for control individuals is shown as the mean ± SD (years).
b Familial indicates the number of patients who are known to have at least one other relative with a similar dementia syndrome.
c Nine of the 297 patients were part of the original FTLD-TDP GWA study (Van Deerlin et al., 2010) and were excluded from the replication analysis.
FTLD-TDP = FTLD pathology with TAR DNA-binding protein inclusions; FTLD-tau = FTLD pathology with MAPT protein inclusion; FTLD-UPS = FTLD pathology with protein inclusions from the ubiquitin proteasome system; FTLD-ni = FTLD pathology with no inclusions of aggregated proteins. NA = not applicable. For more information on the pathological subtypes of FTLD see Mackenzie et al., 2010.
Description of the Flanders–Belgian FTLD patient and control cohort
| Parameters | Flanders-Belgian FTLD cohort | FTLD patient selection for current study | Flanders–Belgian control cohort | Control individual selection for current study |
|---|---|---|---|---|
| N | 315 | 297c | 870 | 595 |
| Age at onset/inclusiona | 62.3 ± 10.6 (29–90) | 62.8 ± 10.7 (29–90) | 65.2 ± 14.9 (19–94) | 61.8 ± 15.4 (19–94) |
| Familialb, n (%) | 119 (37.7%) | 100 (33.7) | NA | NA |
| Mutations, n (%) | 30 (9.5) | 15 (5.1) | NA | NA |
| Pathology diagnoses, n (%) | 22 (7.0) | 14 (4.7) | NA | NA |
| Parameters | Flanders-Belgian FTLD cohort | FTLD patient selection for current study | Flanders–Belgian control cohort | Control individual selection for current study |
|---|---|---|---|---|
| N | 315 | 297c | 870 | 595 |
| Age at onset/inclusiona | 62.3 ± 10.6 (29–90) | 62.8 ± 10.7 (29–90) | 65.2 ± 14.9 (19–94) | 61.8 ± 15.4 (19–94) |
| Familialb, n (%) | 119 (37.7%) | 100 (33.7) | NA | NA |
| Mutations, n (%) | 30 (9.5) | 15 (5.1) | NA | NA |
| Pathology diagnoses, n (%) | 22 (7.0) | 14 (4.7) | NA | NA |
Mutations in the Flanders–Belgian FTLD cohort: 22 GRN (7.0%), 4 MAPT (1.3%), 2 VCP (0.6%), 1 CHMP2B (0.3%) and 1 PSEN1 (0.3%).
Pathology diagnoses in the Flanders–Belgian FTLD cohort: 19 FTLD-TDP (6.0%), 1 FTLD-tau (0.3%), 1 FTLD-UPS (0.3%), 1 FTLD-ni (0.3%).
a Age at onset for patients or age at inclusion for control individuals is shown as the mean ± SD (years).
b Familial indicates the number of patients who are known to have at least one other relative with a similar dementia syndrome.
c Nine of the 297 patients were part of the original FTLD-TDP GWA study (Van Deerlin et al., 2010) and were excluded from the replication analysis.
FTLD-TDP = FTLD pathology with TAR DNA-binding protein inclusions; FTLD-tau = FTLD pathology with MAPT protein inclusion; FTLD-UPS = FTLD pathology with protein inclusions from the ubiquitin proteasome system; FTLD-ni = FTLD pathology with no inclusions of aggregated proteins. NA = not applicable. For more information on the pathological subtypes of FTLD see Mackenzie et al., 2010.
Age at onset analysis was also performed in the Flanders–Belgian GRN founder family, an extended FTLD pedigree segregating the GRN IVS1 + 5G > C null mutation and in which a wide range in onset age was previously reported (Cruts et al., 2006; Brouwers et al., 2007). The founder pedigree consists of 10 branches containing 226 clinically documented individuals, including 41 patients with FTLD and 37 cognitively healthy mutation carriers. Onset age in the patients varied between 45 and 76 years (63.2 ± 7.4 years). One in four mutation carriers was still cognitively intact at age >60 years and three obligate mutation carriers died without clinical signs of FTLD at ages 69, 74 and 81 years. DNA was available for 119 individuals, of whom 21 were patients and 29 unaffected mutation carriers.
Sequencing analysis
Genomic DNA was extracted from whole blood by automated DNA extraction on a Magtration® 12GC PLUS (Precision System Science Co. Ltd, Woerrstadt, Germany). Primers for polymerase chain reaction amplification of genomic DNA and sequencing were designed using ExonPrimer (accessible through the USCS Genome Browser, http://genome.ucsc.edu/, primer sequences will be made available on request). The seven TMEM106B coding exons and non-coding 5′- and 3′-untranslated regions were analysed. Polymerase chain reaction amplicons were purified using the ExoSAP-IT® kit (USB Corporation, Cleveland, OH, USA) and sequenced using the BigDye® Terminator Cycle Sequencing kit v3.1 (Applied Biosystems, Foster City, CA, USA) on an ABI3730 automated sequencer (Applied Biosystems). Sequence variations were detected using the software package novoSNP (Weckx et al., 2005) and confirmed by visual inspection of the DNA sequence traces. The online software tools PMUT (Ferrer-Costa et al., 2005) and FastSNP (Yuan et al., 2006) were used to predict the effect of amino acid substitutions on protein function.
SNP selection and genotyping
To reconstruct the linkage disequilibrium structure in the Flanders–Belgian population, we genotyped 39 SNPs in the control cohort. SNPs were selected from the HapMap2 CEU data and from the common variants detected by sequencing, and were checked for deviations of Hardy-Weinberg equilibrium. For association fine-mapping, nine tag SNPs were selected from within the 36 kb linkage disequilibrium block around TMEM106B plus seven SNPs up- and downstream of the linkage disequilibrium block. SNPs from the HapMap2 CEU data were selected by the pair-wise tagging method with an r2 > 0.8, minor allele frequency >5% and call rate >75%, and complemented by three independent sequencing SNPs to capture the complete genetic variability across the linkage disequilibrium block in the Flanders–Belgian population. All SNPs were genotyped by Sequenom MassArray® technology (Sequenom Inc., Hamburg, Germany). Polymerase chain reaction and extension primers were designed using the Assay Design 3.0 Software (Sequenom Inc., primer sequences will be made available on request). Genotypes were assigned using the TyperAnalyser 4.0 programme (Sequenom Inc.) and checked by visual inspection.
Statistical analyses
With the statistical analysis programme PLINK version 1.07 (Purcell et al., 2007) allelic and genotypic associations were calculated using logistic regression under an additive model taking into account age and gender (age of onset for patients, age at inclusion for control individuals; disease status was set as the dependent variable and genotype as independent variable, age and gender were used as covariables). Onset age analysis for rs10200004 and rs1990622 in the total FTLD patient population was performed by univariate analysis of variances within Statistical Package for the Social Sciences 16.0 (SPSS. Inc., Chicago, Il, USA). In the GRN IVS1 + 5G > C founder family, effect on onset age was estimated in a linkage analysis for censored quantitative traits using a reversible jump Markov chain Monte Carlo algorithm implemented in Loki (Heath, 1997), allowing maximal incorporation of genetic and phenotypic information on all affected and unaffected mutation carriers. Genotype at rs1020004 or rs1990622 for the 21 patients and 29 unaffected carriers was entered in the trait model as a fixed major gene covariate (Wijsman and Yu, 2004).
Gene expression studies
Gene expression analyses were performed in lymphoblast cell lines and brain frontal cortex.
Lymphoblast cell lines from 22 patients and 26 control individuals selected from the Flanders patient–control cohort were analysed. Included in the patient selection were 14 patients with known FTLD-TDP pathology of which nine were GRN and two were VCP mutation carriers, and eight patients had unknown underlying FTLD pathology. Brain frontal cortex samples were obtained from 11 patients with FTLD-TDP including two GRN and three VCP mutation carriers, together with 12 control individuals.
For brain samples, 30–50 mg of fresh-frozen frontal cortex was ground in liquid nitrogen for RNA extraction. Total RNA was extracted using the Ribopure™ kit (Ambion, Applied Biosystems) and treated with DNase (Turbo™ DNase Kit; Ambion, Applied Biosystems). RNA quality was verified on an Agilent 2100 Bioanalyser using the RNA 6000 pico kit (Agilent, Diegm, Belgium) and only samples were included with a RNA integrity number >5.8 (Supplementary Table 3). First strand complementary DNA was synthesized with random hexamer primers using SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). Gene expression was analysed by quantitative reverse-transcription polymerase chain reaction. Amplification was carried out for 40 cycles with TMEM106B specific forward and reverse primers (primer sequences will be made available on request) on an ABI 7900HT instrument (Applied Biosystems) using a SYBR® Green I mastermix (Applied Biosystems). Messenger RNA levels were quantified using the delta–delta CT method and normalized to the geometric mean of the two most stable housekeeping genes (GAPDH and ACTB) from a set of four tested housekeeping genes (GAPDH, SDHA, PPIA and ACTB) as determined by the algorithm GeNorm (Vandesompele et al., 2002). Quantitative reverse-transcription polymerase chain reaction analyses were performed at least in duplicate and each run was followed by a melting curve analysis to ensure that only one product was formed at the expected melting temperature. Differences in TMEM106B expression were analysed by non-parametric Kruskal–Wallis test for the comparison of three groups and Mann–Whitney U-test for two group comparisons.
Results
Replication of TMEM106B locus association
The first aim in our study was to replicate association with the three genome-wide associated SNPs at 7p21.3 in an independent sample of primarily clinical diagnosed FTLD patients (n = 288) and control individuals (n = 595), ascertained in Flanders–Belgium. Allelic association reached significant values of P = 0.008, 0.013 and 0.041 for rs1990622, rs6966915 and rs1020004, respectively (Table 2). For rs1990622, showing the strongest association, the odds ratio for the minor C-allele was 0.75 (95% confidence interval 0.61–0.93), which corresponds to an increased risk associated with the common allele of 1.33. This positive replication validated further in-depth analyses of TMEM106B as a novel risk factor for FTLD.
Allelic association of the FTLD-TDP genome-wide association study SNPs at the TMEM106B locus in the Flanders–Belgian FTLD cohort
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.79 (0.63–0.99) | 0.041 |
| rs6966915 | T | 0.35 | 0.42 | 1.00 | 0.77 (0.62–0.95) | 0.013 |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.75 (0.61–0.93) | 0.008 |
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.79 (0.63–0.99) | 0.041 |
| rs6966915 | T | 0.35 | 0.42 | 1.00 | 0.77 (0.62–0.95) | 0.013 |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.75 (0.61–0.93) | 0.008 |
SNPs are listed according to their genomic order on chromosome 7. Minor allele frequencies are given for 288 patients not previously included in the original genome-wide association study (Van Deerlin et al., 2010) and for 595 control individuals. P-values and odds ratio (OR) with associated 95% confidence interval (CI) were calculated under an additive model using logistic regression models adjusted for age and gender.
HWE = P-value for the test of Hardy–Weinberg equilibrium in control individuals.
Allelic association of the FTLD-TDP genome-wide association study SNPs at the TMEM106B locus in the Flanders–Belgian FTLD cohort
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.79 (0.63–0.99) | 0.041 |
| rs6966915 | T | 0.35 | 0.42 | 1.00 | 0.77 (0.62–0.95) | 0.013 |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.75 (0.61–0.93) | 0.008 |
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.79 (0.63–0.99) | 0.041 |
| rs6966915 | T | 0.35 | 0.42 | 1.00 | 0.77 (0.62–0.95) | 0.013 |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.75 (0.61–0.93) | 0.008 |
SNPs are listed according to their genomic order on chromosome 7. Minor allele frequencies are given for 288 patients not previously included in the original genome-wide association study (Van Deerlin et al., 2010) and for 595 control individuals. P-values and odds ratio (OR) with associated 95% confidence interval (CI) were calculated under an additive model using logistic regression models adjusted for age and gender.
HWE = P-value for the test of Hardy–Weinberg equilibrium in control individuals.
Sequencing analysis
To map the genetic variability at the TMEM106B locus and to look for potential pathogenic mutations, we sequenced all coding exons, 3′- and 5′-untranslated region exons and ∼100 bps of adjacent intronic regions. Direct sequencing revealed a total of 61 variations, 40 common SNPs with a minor allele frequency >0.05 (Supplementary Table 1) and 21 rare SNPs (minor allele frequency ≤0.05, Supplementary Table 2). Two SNPs predicted amino acid substitutions, S134N and T185S. S134N is located in exon 5 and was detected with equal frequencies of 2% in patients as in control individuals (P = 0.601). T185S or rs3173615 is located in exon 6, and was observed with a frequency of 35% in patients and 42% in control individuals (P = 0.003). Of the remaining SNPs, none were predicted to be near splice-sites, transcription factor binding sites, or microRNA binding sites (UCSC Genome Browser). Considering the results of the mutation analysis, it appears that TMEM106B does not bear highly penetrant mutations associated with FTLD in our patient cohort.
Association fine-mapping
The linkage disequilibrium pattern in the European and in our Flanders–Belgian population delineated a 36 kb linkage disequilibrium block containing only the gene TMEM106B (Fig. 1). Nine tag SNPs were selected from within the linkage disequilibrium block to capture its complete genetic variability in the Flanders–Belgian population. In addition, four independent SNPs were chosen upstream and three downstream of the linkage disequilibrium block for further association testing in the 297 patients with FTLD and 595 control individuals.

Linkage disequilibrium pattern at the TMEM106B locus. Linkage disequilibrium plot for Hapmap2 CEU data, D′/LOD colour scheme, interval 12.200–12.270 kb. The linkage disequilibrium structure delineates a 36 kb block including only the TMEM106B gene. The relative position of the three replicated SNPs from the genome-wide association study are indicated by ‘X’.
Single-SNP allelic association testing achieved significant P-values (P < 0.05) for the replicated genome-wide association study tag SNPs as well as for one more SNP within the linkage disequilibrium block (Table 3, minimum P-value of 0.003 for rs1990622). After calculation of genotype frequencies and associated effect sizes, only the two genome-wide association study SNPs rs1020004 and rs1990622, sustained significance (Table 4). A minimal odds ratio was calculated for rs1020004 [odds ratioCC = 0.49, (95% confidence interval = 0.27–0.87), P = 0.014]. For rs1990622, one copy of the risk allele was sufficient to achieve a significantly decreased risk effect [odds ratioCT = 0.69, (95% confidence interval 0.51–0.93), P = 0.017; odds ratioCC = 0.55, (95% confidence interval 0.36–0.86), P = 0.009]. In the Flanders–Belgian population, correlation between rs1020004 and rs1990622 was 79%.
Allelic association of tag SNPs at the TMEM106B locus in the Flanders–Belgian FTLD cohort
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs4719302 | G | 0.49 | 0.46 | 0.673 | – | ns |
| rs13244354 | C | 0.10 | 0.12 | 0.564 | ns | |
| rs17165706 | T | 0.24 | 0.22 | 1.000 | – | ns |
| rs1435528 | G | 0.34 | 0.32 | 0.153 | – | ns |
| rs1865567 | G | 0.16 | 0.21 | 1.00 | 0.74 (0.55–0.99) | 0.039 |
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.77 (0.62–0.97) | 0.024 |
| rs17165746 | A | 0.08 | 0.1 | 0.495 | – | ns |
| rs13234238 | C | 0.08 | 0.09 | 0.313 | – | ns |
| rs1047601 | T | 0.16 | 0.13 | 0.85 | – | ns |
| rs10488192 | T | 0.16 | 0.19 | 0.682 | – | ns |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.73 (0.59–0.90) | 0.003 |
| rs1548882 | C | 0.5 | 0.47 | 0.352 | – | ns |
| rs6945902 | A | 0.2 | 0.21 | 0.219 | – | ns |
| rs6952078 | T | 0.09 | 0.11 | 0.397 | – | ns |
| rs11977828 | T | 0.26 | 0.26 | 0.387 | – | ns |
| rs7457114 | A | 0.26 | 0.23 | 0.403 | – | ns |
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs4719302 | G | 0.49 | 0.46 | 0.673 | – | ns |
| rs13244354 | C | 0.10 | 0.12 | 0.564 | ns | |
| rs17165706 | T | 0.24 | 0.22 | 1.000 | – | ns |
| rs1435528 | G | 0.34 | 0.32 | 0.153 | – | ns |
| rs1865567 | G | 0.16 | 0.21 | 1.00 | 0.74 (0.55–0.99) | 0.039 |
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.77 (0.62–0.97) | 0.024 |
| rs17165746 | A | 0.08 | 0.1 | 0.495 | – | ns |
| rs13234238 | C | 0.08 | 0.09 | 0.313 | – | ns |
| rs1047601 | T | 0.16 | 0.13 | 0.85 | – | ns |
| rs10488192 | T | 0.16 | 0.19 | 0.682 | – | ns |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.73 (0.59–0.90) | 0.003 |
| rs1548882 | C | 0.5 | 0.47 | 0.352 | – | ns |
| rs6945902 | A | 0.2 | 0.21 | 0.219 | – | ns |
| rs6952078 | T | 0.09 | 0.11 | 0.397 | – | ns |
| rs11977828 | T | 0.26 | 0.26 | 0.387 | – | ns |
| rs7457114 | A | 0.26 | 0.23 | 0.403 | – | ns |
SNPs are listed according to their genomic order on chromosome 7. SNPs from Row 5 to Row 13 correspond to the nine tag SNPs located within the 36 kb linkage disequilibrium block. SNPs in bold are the replicated tag SNPs from the FTLD-TDP genome-wide association study (Van Deerlin et al., 2010). Minor allele frequencies in 297 patients and 595 control individuals are given. P-values and odds ratio (OR) with associated 95% confidence interval (CI) were calculated under an additive model using logistic regression models adjusted for age and gender. Results for SNPs with significant P < 0.05 are listed.
HWE = P-value for the test of Hardy–Weinberg equilibrium in control individuals; ns = not significant.
Allelic association of tag SNPs at the TMEM106B locus in the Flanders–Belgian FTLD cohort
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs4719302 | G | 0.49 | 0.46 | 0.673 | – | ns |
| rs13244354 | C | 0.10 | 0.12 | 0.564 | ns | |
| rs17165706 | T | 0.24 | 0.22 | 1.000 | – | ns |
| rs1435528 | G | 0.34 | 0.32 | 0.153 | – | ns |
| rs1865567 | G | 0.16 | 0.21 | 1.00 | 0.74 (0.55–0.99) | 0.039 |
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.77 (0.62–0.97) | 0.024 |
| rs17165746 | A | 0.08 | 0.1 | 0.495 | – | ns |
| rs13234238 | C | 0.08 | 0.09 | 0.313 | – | ns |
| rs1047601 | T | 0.16 | 0.13 | 0.85 | – | ns |
| rs10488192 | T | 0.16 | 0.19 | 0.682 | – | ns |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.73 (0.59–0.90) | 0.003 |
| rs1548882 | C | 0.5 | 0.47 | 0.352 | – | ns |
| rs6945902 | A | 0.2 | 0.21 | 0.219 | – | ns |
| rs6952078 | T | 0.09 | 0.11 | 0.397 | – | ns |
| rs11977828 | T | 0.26 | 0.26 | 0.387 | – | ns |
| rs7457114 | A | 0.26 | 0.23 | 0.403 | – | ns |
| dbSNP ID | Minor allele | Minor allele frequency | HWE | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|
| Patients | Controls | |||||
| rs4719302 | G | 0.49 | 0.46 | 0.673 | – | ns |
| rs13244354 | C | 0.10 | 0.12 | 0.564 | ns | |
| rs17165706 | T | 0.24 | 0.22 | 1.000 | – | ns |
| rs1435528 | G | 0.34 | 0.32 | 0.153 | – | ns |
| rs1865567 | G | 0.16 | 0.21 | 1.00 | 0.74 (0.55–0.99) | 0.039 |
| rs1020004 | C | 0.26 | 0.31 | 0.44 | 0.77 (0.62–0.97) | 0.024 |
| rs17165746 | A | 0.08 | 0.1 | 0.495 | – | ns |
| rs13234238 | C | 0.08 | 0.09 | 0.313 | – | ns |
| rs1047601 | T | 0.16 | 0.13 | 0.85 | – | ns |
| rs10488192 | T | 0.16 | 0.19 | 0.682 | – | ns |
| rs1990622 | C | 0.35 | 0.42 | 1.00 | 0.73 (0.59–0.90) | 0.003 |
| rs1548882 | C | 0.5 | 0.47 | 0.352 | – | ns |
| rs6945902 | A | 0.2 | 0.21 | 0.219 | – | ns |
| rs6952078 | T | 0.09 | 0.11 | 0.397 | – | ns |
| rs11977828 | T | 0.26 | 0.26 | 0.387 | – | ns |
| rs7457114 | A | 0.26 | 0.23 | 0.403 | – | ns |
SNPs are listed according to their genomic order on chromosome 7. SNPs from Row 5 to Row 13 correspond to the nine tag SNPs located within the 36 kb linkage disequilibrium block. SNPs in bold are the replicated tag SNPs from the FTLD-TDP genome-wide association study (Van Deerlin et al., 2010). Minor allele frequencies in 297 patients and 595 control individuals are given. P-values and odds ratio (OR) with associated 95% confidence interval (CI) were calculated under an additive model using logistic regression models adjusted for age and gender. Results for SNPs with significant P < 0.05 are listed.
HWE = P-value for the test of Hardy–Weinberg equilibrium in control individuals; ns = not significant.
Genotypic association at the TMEM106B locus in the Flanders–Belgian FTLD cohort
| dbSNP ID | Genotype | Genotype frequency | OR (95% CI) | P-value | |
|---|---|---|---|---|---|
| Patients | Controls | ||||
| rs1865567 | AA | 0.70 | 0.63 | – | – |
| AG | 0.28 | 0.33 | 0.78 (0.56–1.10) | 0.151 | |
| GG | 0.00 | 0.04 | 0.40 (0.14–1.18) | 0.096 | |
| rs1020004 | TT | 0.54 | 0.48 | – | – |
| TC | 0.41 | 0.42 | 0.87 (0.65–1.17) | 0.362 | |
| CC | 0.06 | 0.10 | 0.49 (0.27–0.87) | 0.014 | |
| rs1990622 | TT | 0.43 | 0.34 | – | – |
| TC | 0.44 | 0.49 | 0.69 (0.51–0.93) | 0.017 | |
| CC | 0.13 | 0.18 | 0.55 (0.36–0.86) | 0.009 | |
| dbSNP ID | Genotype | Genotype frequency | OR (95% CI) | P-value | |
|---|---|---|---|---|---|
| Patients | Controls | ||||
| rs1865567 | AA | 0.70 | 0.63 | – | – |
| AG | 0.28 | 0.33 | 0.78 (0.56–1.10) | 0.151 | |
| GG | 0.00 | 0.04 | 0.40 (0.14–1.18) | 0.096 | |
| rs1020004 | TT | 0.54 | 0.48 | – | – |
| TC | 0.41 | 0.42 | 0.87 (0.65–1.17) | 0.362 | |
| CC | 0.06 | 0.10 | 0.49 (0.27–0.87) | 0.014 | |
| rs1990622 | TT | 0.43 | 0.34 | – | – |
| TC | 0.44 | 0.49 | 0.69 (0.51–0.93) | 0.017 | |
| CC | 0.13 | 0.18 | 0.55 (0.36–0.86) | 0.009 | |
Of the SNPs showing significant allelic association, genotype frequencies in 297 patients and 595 control individuals are given. SNPs in bold are the replicated SNPs form the FTLD-TDP genome-wide association study (Van Deerlin et al., 2010). P-values and odds ratio (OR) with associated 95% confidence interval (CI) were calculated under an additive model using logistic regression models adjusted for age and gender using the common genotype as reference. Significant P-values are in bold.
Genotypic association at the TMEM106B locus in the Flanders–Belgian FTLD cohort
| dbSNP ID | Genotype | Genotype frequency | OR (95% CI) | P-value | |
|---|---|---|---|---|---|
| Patients | Controls | ||||
| rs1865567 | AA | 0.70 | 0.63 | – | – |
| AG | 0.28 | 0.33 | 0.78 (0.56–1.10) | 0.151 | |
| GG | 0.00 | 0.04 | 0.40 (0.14–1.18) | 0.096 | |
| rs1020004 | TT | 0.54 | 0.48 | – | – |
| TC | 0.41 | 0.42 | 0.87 (0.65–1.17) | 0.362 | |
| CC | 0.06 | 0.10 | 0.49 (0.27–0.87) | 0.014 | |
| rs1990622 | TT | 0.43 | 0.34 | – | – |
| TC | 0.44 | 0.49 | 0.69 (0.51–0.93) | 0.017 | |
| CC | 0.13 | 0.18 | 0.55 (0.36–0.86) | 0.009 | |
| dbSNP ID | Genotype | Genotype frequency | OR (95% CI) | P-value | |
|---|---|---|---|---|---|
| Patients | Controls | ||||
| rs1865567 | AA | 0.70 | 0.63 | – | – |
| AG | 0.28 | 0.33 | 0.78 (0.56–1.10) | 0.151 | |
| GG | 0.00 | 0.04 | 0.40 (0.14–1.18) | 0.096 | |
| rs1020004 | TT | 0.54 | 0.48 | – | – |
| TC | 0.41 | 0.42 | 0.87 (0.65–1.17) | 0.362 | |
| CC | 0.06 | 0.10 | 0.49 (0.27–0.87) | 0.014 | |
| rs1990622 | TT | 0.43 | 0.34 | – | – |
| TC | 0.44 | 0.49 | 0.69 (0.51–0.93) | 0.017 | |
| CC | 0.13 | 0.18 | 0.55 (0.36–0.86) | 0.009 | |
Of the SNPs showing significant allelic association, genotype frequencies in 297 patients and 595 control individuals are given. SNPs in bold are the replicated SNPs form the FTLD-TDP genome-wide association study (Van Deerlin et al., 2010). P-values and odds ratio (OR) with associated 95% confidence interval (CI) were calculated under an additive model using logistic regression models adjusted for age and gender using the common genotype as reference. Significant P-values are in bold.
SNP rs1990622 tags 27 additional markers with a high degree of linkage disequilibrium (r2 range 0.93–1). These 27 SNPs include the third SNP from the genome-wide association study, rs6966915, but also rs3173615, which is the non-synonymous SNP coding for T185S that we observed in the sequencing analysis. SNP rs1020004 captures three more SNPs (r2 of 1, 1 and 0.88).
Onset age analysis
Analysis of onset age correlation in the Flanders–Belgian FTLD cohort did not demonstrate an effect of rs1020004 or rs1990622. Mean difference in onset ages between carriers of the CT genotype (one copy of the protective allele) and TT genotype was −2.5 years for rs1020004 (P = 0.08) and −2.0 years for rs1990622 (P = 0.171). Mean differences in onset ages between carriers of the CC genotype (two copies of the protective allele) and TT genotype was −1.8 years for rs1020004 (P = 0.53) and −0.3 years for rs1990622 (P = 0.88). Likewise in the Flanders–Belgian GRN IVS1 + 5G > C founder family, the TMEM106B SNPs could not explain the observed wide range in onset age in GRN mutation carriers. Mean difference in onset ages between CT and TT genotype carriers was −0.39 years for rs1020004 (P = 0.40, minor allele frequency = 0.23), and 1.9 years for rs1990622 (P = 0.20, minor allele frequency = 0.30). Mean difference in onset ages between CC and TT genotype carriers was 1.0 years for rs1020004 (P = 0.50) and −0.6 years for rs1990622 (P = 0.50).
Gene expression studies
Correlation of TMEM106B messenger RNA expression with rs1990622 and disease status was evaluated in lymphoblast cell lines and brain frontal cortex (Fig. 2). In lymphoblasts, quantification of TMEM106B messenger RNA levels in patients demonstrated no differential expression compared with control individuals (P = 0.508, Fig. 2A). To evaluate a possible effect of rs1990622 genotype, data of patients and control individuals were pooled to maximize the sample size and analysed per genotype group; however, comparison of expression yielded no significant correlation (PCC versus CT versus TT = 0.484, Fig. 2B). In patients only or controls only, correlation with genotype was also negative (data not shown). Similarly, in brain frontal cortex, we did not observe increased TMEM106B expression. Mean messenger RNA levels in patients with FTLD-TDP (n = 11) was comparable to that in control individuals (n = 12, P = 0.806) as were the levels in the two genotype groups (PCT versus TT = 0.841, no high quality brain RNA samples were available for CC carriers).
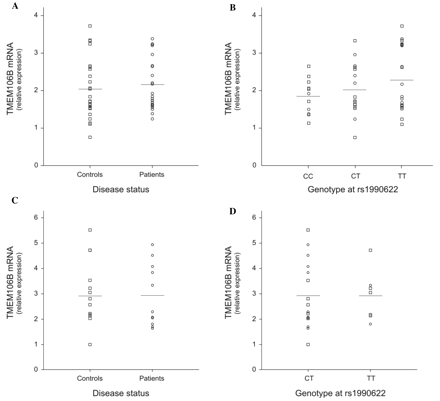
TMEM106B messenger RNA levels in lymphoblast cell lines and brain frontal cortex by disease status and genotype. TMEM106B messenger RNA expression was measured by quantitative reverse-transcription polymerase chain reaction. (A) Relative gene expression in lymphoblast cell lines from patients (n = 22) compared with control individuals (n = 26). (B) Relative gene expression in lymphoblast cell lines from patients and control individuals pooled according to genotype at rs1990622. (C) Relative gene expression in brain frontal cortex from FTLD-TDP patients (n = 11) compared with control individuals (n = 12). (D) Relative gene expression in brain frontal cortex from patients and control individuals pooled according to genotype at rs1990622. In general, we found no evidence that TMEM106B messenger RNA expression is increased in FTLD patients or that TMEM106B rs1990622 genotype is correlated with gene expression. Circles = patients; squares = controls; horizontal lines = group mean.
Discussion
We performed a replication study of the TMEM106B genome-wide association in FTLD-TDP. Whereas the original genome-wide association study exclusively included patients with autopsy confirmed TDP pathology or with a GRN mutation with predicted TDP pathology (Van Deerlin et al., 2010), our cohort consisted of patients with primarily clinically diagnosed FTLD. To minimize genetic heterogeneity, we excluded autopsy confirmed patients without TDP pathology and patients carrying a MAPT or CHMP2B mutation, which are known as non-TDP pathologies (Table 1). Sampling of patients and control individuals in the small geographical region of Flanders in Belgium further favoured the genetic homogeneity of our study population.
We replicated association with the three TMEM106B SNPs from the FTLD-TDP genome-wide association study and demonstrated that the association is probably restricted to a 36 kb genomic region containing only the gene TMEM106B. The TMEM106B SNPs, rs1990622 and rs1020004 were the most strongly associated independent SNPs in the associated region. Carriers of one or two copies of the TMEM106B protective alleles have a 30–50% reduced risk of developing FTLD. Although we did not identify other independent associated SNPs, rs1990622 and rs1020004 were in high linkage disequilibrium with 30 more SNPs. The functional relevance of these SNPs to FTLD remains unknown at present. However, the fact that they are all located in or very close to TMEM106B, and that no other genes were present in or near the associated region (Fig. 1), makes TMEM106B the most likely candidate underlying the FTLD susceptibility. In silico prediction of the functional properties of the 32 SNPs anticipated the highest impact for rs3173615, a non-synonymous SNP coding for a threonine (T) to serine (S) substitution, though the predicted effect on protein function was modest. The missense mutation is positioned at codon 185, C-terminal of the transmembrane domain (codon 97–117). Except for being a single-pass transmembrane protein, little is currently known about the function of TMEM106B, making it difficult to estimate the potential impact of amino acid substitutions on its biological function. Analysis of protein conservation however indicated that the T-residue at position 185 is highly conserved among vertebrates.
In the original FTLD-TDP genome-wide association study, association with 7p21.3 was most pronounced in the subset of patients carrying a GRN mutation. Based on this finding, it was hypothesized that TMEM160B might act as a disease modifier in GRN-associated FTLD. We investigated a putative correlation of rs1040002 and rs1990622 with onset age in the Flanders–Belgian FTLD cohort but found no evidence for association. Also, in an extended Flanders–Belgian FTLD founder family, characterized by wide intrafamilial variability in onset age (Cruts et al., 2006; Brouwers et al., 2007), the TMEM106B risk variants failed to explain the >30 years onset age range in GRN IVS1 + 5G > C carriers.
Further, we did not find evidence that TMEM106B expression correlated with disease. Although expression in Epstein-Barr virus cell lines is known to be variable (Choy et al., 2008), the use of lymphoblast cell lines allows the analysis of relatively larger samples sizes. In contrast, obtaining high-quality RNA from a significant number of brain samples is challenging. In our study we were able to analyse brain samples from 11 patients with FTLD-TDP and 12 control individuals. In this sample set we observed no increased TMEM106B expression in patients with FTLD-TDP compared with controls. Although our number of brain samples was limited, it was in the same range as the original study who found a positive correlation with TMEM106B expression in 25 brains (Van Deerlin et al., 2010). Expression analyses of larger number of patient and control brains will be vital to resolve whether or not TMEM106B pathogenesis is mediated by variable gene expression.
Taken together, our study strongly supports TMEM106B as a risk gene for FTLD though further research is needed to learn whether T185S, variable gene expression or another mechanism contributes to the disease biology driving the association of TMEM106B with FTLD. As other replication studies of TMEM106B are being reported in geographically distinct populations, the extent of the genetic contribution of TMEM106B to FTLD risk will become clear. Interestingly, while our paper was under review, the principal authors of the FTLD-TDP genome wide association study published their observations that TMEM106B genotypes also seem to influence the development of cognitive impairment in ALS patients, further broadening the implications of the TDP-43 proteinopathy spectrum that binds FTLD and ALS (Vass et al., 2010).
Funding
The research described by the authors was funded in part by the Interuniversity Attraction Poles (IAP) programme P6/43 of the Belgian Federal Science Policy office; the Foundation for Alzheimer Research (SAO/FRMA); the Association for Frontotemporal Dementias (AFTD); the Medical Foundation Queen Elisabeth; a Methusalem excellence grant of the Flemish Government; the Research Foundation Flanders (FWO); the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-V); the Special Research Fund (BOF) of the University of Antwerp, Belgium; the IWT-V provided a PhD fellowship (to T.V.L.); the FWO provided a PhD fellowship (to G.K.), a post-doctoral fellowship (to K.S. and J.v.d.Z.); a senior clinical investigator mandate (to R.V.).
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
The authors are grateful to the participants to this study for their kind co-operation. We further acknowledge the contribution of the personnel of the VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be), and of the Antwerp Biobank of the Institute Born-Bunge (IBB).


{kind=link}
{kind=link}