




Abstract
Frontotemporal dementia and amyotrophic lateral sclerosis are closely related clinical syndromes with overlapping molecular pathogenesis. Several families have been reported with members affected by frontotemporal dementia, amyotrophic lateral sclerosis or both, which show genetic linkage to a region on chromosome 9p21. Recently, two studies identified the FTD/ALS gene defect on chromosome 9p as an expanded GGGGCC hexanucleotide repeat in a non-coding region of the chromosome 9 open reading frame 72 gene (C9ORF72). In the present study, we provide detailed analysis of the clinical features and neuropathology for 16 unrelated families with frontotemporal dementia caused by the C9ORF72 mutation. All had an autosomal dominant pattern of inheritance. Eight families had a combination of frontotemporal dementia and amyotrophic lateral sclerosis while the other eight had a pure frontotemporal dementia phenotype. Clinical information was available for 30 affected members of the 16 families. There was wide variation in age of onset (mean = 54.3, range = 34–74 years) and disease duration (mean = 5.3, range = 1–16 years). Early diagnoses included behavioural variant frontotemporal dementia (n = 15), progressive non-fluent aphasia (n = 5), amyotrophic lateral sclerosis (n = 9) and progressive non-fluent aphasia–amyotrophic lateral sclerosis (n = 1). Heterogeneity in clinical presentation was also common within families. However, there was a tendency for the phenotypes to converge with disease progression; seven subjects had final clinical diagnoses of both frontotemporal dementia and amyotrophic lateral sclerosis and all of those with an initial progressive non-fluent aphasia diagnosis subsequently developed significant behavioural abnormalities. Twenty-one affected family members came to autopsy and all were found to have transactive response DNA binding protein with Mr 43 kD (TDP-43) pathology in a wide neuroanatomical distribution. All had involvement of the extramotor neocortex and hippocampus (frontotemporal lobar degeneration-TDP) and all but one case (clinically pure frontotemporal dementia) had involvement of lower motor neurons, characteristic of amyotrophic lateral sclerosis. In addition, a consistent and relatively specific pathological finding was the presence of neuronal inclusions in the cerebellar cortex that were ubiquitin/p62-positive but TDP-43-negative. Our findings indicate that the C9ORF72 mutation is a major cause of familial frontotemporal dementia with TDP-43 pathology, that likely accounts for the majority of families with combined frontotemporal dementia/amyotrophic lateral sclerosis presentation, and further support the concept that frontotemporal dementia and amyotrophic lateral sclerosis represent a clinicopathological spectrum of disease with overlapping molecular pathogenesis.
Introduction
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are closely related clinical syndromes with overlapping molecular pathogenesis (Mackenzie et al., 2010). A significant proportion of patients with FTD develop features of motor neuron dysfunction (Lomen-Hoerth et al., 2002; Burrell et al., 2011) and up to half of those presenting with ALS are found to have evidence of frontal lobe impairment (Lomen-Hoerth et al., 2003; Phukan et al., 2007). The concept that FTD and ALS represent a clinicopathological spectrum of disease is strongly supported by the discovery of the transactive response DNA binding protein with Mr 43 kD (TDP-43) as the pathological protein in the vast majority of cases with ALS and in the most common pathological subtype of FTD (Neumann et al., 2006; Cairns et al., 2007; Mackenzie et al., 2007), now referred to as frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP) (Mackenzie et al., 2009).
Both FTD and ALS have a strong genetic basis (Valdmanis et al., 2009; See et al., 2010). Most of the known gene abnormalities preferentially cause just one phenotype; mutations in the microtubule associated protein tau (MAPT) and progranulin (GRN) genes are major causes of familial FTD but not motor neuron disease, while mutations in the Cu/Zn superoxide dismutase 1 (SOD1), TDP-43 (TARDBP) and fused in sarcoma (FUS) genes tend to cause familial ALS without dementia. However, a number of families have been reported with an autosomal dominant pattern of disease in which affected members may develop either FTD or ALS or both (FTD–ALS). Several of these families have shown genetic linkage to a region on chromosome 9p21, with the combined data defining a minimum linkage region of 3.7 Mb, containing only five known genes (Momeni et al., 2006; Morita et al., 2006; Vance et al., 2006; Valdmanis et al., 2007; Luty et al., 2008; Le Ber et al., 2009; Gijselinck et al., 2010; Boxer et al., 2011; Pearson et al., 2011). Importantly, the same chromosomal region has been identified in several large independent genome-wide association studies of both ALS and FTD, implicating the genetic defect at chromosome 9p in sporadic forms of both diseases (van Es et al., 2009; Laaksovirta et al., 2010; Shatunov et al., 2010; van Deerlin et al., 2010).
Recently, two independent studies reported identification of the FTD/ALS gene defect on chromosome 9p as being a massively expanded GGGGCC hexanucleotide repeat in a non-coding region of the chromosome 9 open reading frame 72 gene (C9ORF72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The mutation was found to result in loss of one alternatively spliced C9ORF72 transcript and the formation of nuclear RNA foci composed of the hexanucleotide repeat, suggesting multiple pathogenic mechanisms (DeJesus-Hernandez et al., 2011). In these two studies, the C9ORF72 mutation was found to be the most common genetic abnormality in familial and sporadic forms of both FTD and of ALS and was particularly frequent in patients and families with both conditions. However, the descriptions of the associated clinical features were limited to basic demographic information and broad phenotypes, while the neuropathology was described for only one family (DeJesus-Hernandez et al., 2011).
In the present study, we provide a detailed analysis of the clinical features and neuropathology for 16 unrelated families with FTD (±ALS) caused by the C9ORF72 mutation. We highlight the degree of clinical heterogeneity and specific pathological changes associated with this important, newly discovered genetic abnormality.
Materials and methods
Subjects
The subjects included in this study were part of a series screened for the C9ORF72 mutation in one of the recent mutation discovery studies (DeJesus-Hernandez et al., 2011). All subjects were participants in an ongoing FTD research study at the University of British Columbia (Vancouver, Canada), which focuses on the longitudinal assessment of relatives of probands with pathologically confirmed FTLD-TDP. The initial cohort consisted of 29 unrelated probands with autopsy proven FTLD-TDP (Table 1). Twenty-three probands had a family history suggestive of autosomal dominant inheritance with multiple affected family members. Three others were apparently sporadic and three had only a single first-degree relative with clinical dementia, not fulfilling FTD criteria and without autopsy confirmation (classified as ‘possible family history’). All probands (n = 29) had previously been screened for GRN mutations and seven were found to be positive. As part of the recent gene discovery study, DNA samples from the remaining 22 GRN mutation-negative probands (or one of their affected relatives) were analysed using a combination of fluorescent fragment-length and repeat-primed polymerase chain reaction analyses, and 16 were found to carry alleles with abnormally expanded GGGGCC hexanucleotide repeats in intron 1 of C9ORF72. While detailed molecular genetic methodology and basic demographic features of the probands have been published previously (DeJesus-Hernanadez et al., 2011), the current study focuses on the detailed analysis of the clinical and pathological features of these 16 families with FTLD-TDP caused by the C9ORF72 mutation.
Mutations in probandsa with autopsy proven FTLD-TDP pathology
| Inheritance | Phenotype | n | Mutation | |
|---|---|---|---|---|
| C9ORF72 | GRN | |||
| Autosomal dominant | FTD/ALS | 8 | 8 | 0 |
| Autosomal dominant | FTD | 15 | 8 | 7 |
| Possible family history | FTD/ALS | 1 | 0 | 0 |
| Possible family history | FTD | 2 | 0 | 0 |
| Sporadic | FTD/ALS | 1 | 0 | 0 |
| Sporadic | FTD | 2 | 0 | 0 |
| Total | 29 | 16 | 7 | |
| Inheritance | Phenotype | n | Mutation | |
|---|---|---|---|---|
| C9ORF72 | GRN | |||
| Autosomal dominant | FTD/ALS | 8 | 8 | 0 |
| Autosomal dominant | FTD | 15 | 8 | 7 |
| Possible family history | FTD/ALS | 1 | 0 | 0 |
| Possible family history | FTD | 2 | 0 | 0 |
| Sporadic | FTD/ALS | 1 | 0 | 0 |
| Sporadic | FTD | 2 | 0 | 0 |
| Total | 29 | 16 | 7 | |
a In some cases, DNA was not available from the FTLD-TDP-positive proband and the mutation was identified in an affected first-degree relative. Possible family history = one first-degree relative with dementia of unspecified type.
GRN = progranulin gene.
Mutations in probandsa with autopsy proven FTLD-TDP pathology
| Inheritance | Phenotype | n | Mutation | |
|---|---|---|---|---|
| C9ORF72 | GRN | |||
| Autosomal dominant | FTD/ALS | 8 | 8 | 0 |
| Autosomal dominant | FTD | 15 | 8 | 7 |
| Possible family history | FTD/ALS | 1 | 0 | 0 |
| Possible family history | FTD | 2 | 0 | 0 |
| Sporadic | FTD/ALS | 1 | 0 | 0 |
| Sporadic | FTD | 2 | 0 | 0 |
| Total | 29 | 16 | 7 | |
| Inheritance | Phenotype | n | Mutation | |
|---|---|---|---|---|
| C9ORF72 | GRN | |||
| Autosomal dominant | FTD/ALS | 8 | 8 | 0 |
| Autosomal dominant | FTD | 15 | 8 | 7 |
| Possible family history | FTD/ALS | 1 | 0 | 0 |
| Possible family history | FTD | 2 | 0 | 0 |
| Sporadic | FTD/ALS | 1 | 0 | 0 |
| Sporadic | FTD | 2 | 0 | 0 |
| Total | 29 | 16 | 7 | |
a In some cases, DNA was not available from the FTLD-TDP-positive proband and the mutation was identified in an affected first-degree relative. Possible family history = one first-degree relative with dementia of unspecified type.
GRN = progranulin gene.
Clinical information
All available clinical records of 30 affected members from the 16 families with the C9ORF72 mutation were reviewed. This included retrospective chart review for deceased individuals and the evaluation of clinical data collected prospectively during the longitudinal assessment of living subjects. Clinical features were scored using a semi-quantitative grading system (0, absent; 1, mild; 2, moderate; 3, severe). The amount and quality of available clinical information varied considerably among subjects and features that could not be evaluated with confidence were not scored. A clinical diagnosis of FTD was based on Neary criteria (Neary et al., 1998), while ALS diagnosis was based on El Escorial criteria (Brooks et al., 2000). One of the families (Family E) has previously been described in detail (Boxer et al., 2010). For this family, only the proband was included in the present analysis.
Neuropathology
In addition to the 16 probands, post-mortem material was available from an additional five affected family members (total 21 autopsies). Twenty of these cases were also included in the clinical evaluation (above). Pathological changes were evaluated in multiple brain and spinal cord regions and graded using a semi-quantitative system (0, absent; 1, mild; 2, moderated; 3, severe). Sections of cerebellum from 25 cases in which the C9ORF72 had been excluded (sporadic ALS, n = 10; familial FTD with GRN mutations, n = 10; sporadic FTLD-TDP, n = 5) were evaluated to determine the specificity of ubiquitin/p62-immunoreactive cerebellar inclusions. Controls for C9ORF72 immunohistochemistry included hippocampal sections from cases of FTLD-TDP without C9ORF72 mutation, FTLD-tau, FTLD-FUS, Alzheimer's disease and spinal cord sections from cases of sporadic ALS and ALS with SOD1 mutations (n = 5, each).
Histochemistry and immunohistochemistry
Sections of formalin fixed, paraffin-embedded tissue were stained with haematoxylin and eosin, haematoxylin and eosin combined with Luxol fast blue and Bielschowsky silver method. Immunohistochemistry was performed on 5-µm thick sections using the Ventana BenchMark® XT automated staining system (Ventana) and developed with aminoethylcarbizole. The primary antibodies employed recognized ubiquitin (Dako anti-ubiquitin; 1:500, following microwave antigen retrieval), p62 (BD Transduction Laboratories p62 Lck ligand; 1:500 following microwave antigen retrieval), hyperphosphorylated tau (Innogenetics AT-8; 1:2000 following microwave antigen retrieval and Sigma TAU-2; 1:1000 with 3 h initial incubation at room temperature), α-synuclein (Zymed anti-α-synuclein; 1:10 000, following microwave antigen retrieval), Aβ (Dako anti-beta amyloid; 1:100 with initial incubation for 3 h at room temperature), TDP-43 (ProteinTech Group anti-TARDBP; 1:1000 following microwave antigen retrieval), FUS (Sigma-Aldrich anti-FUS, 1:200, initial overnight incubation at room temperature, following microwave antigen retrieval) and C9ORF72 (Sigma-Aldrich, anti-C9orf72; 1:50 overnight incubation following microwave antigen retrieval).
Statistical analysis
Descriptive statistics was used to characterize the demographics of the cohort. Age of onset and duration of disease were compared between subjects with and without ALS by t-test. ANOVA with post hoc Student–Newman–Keuls test was used for multiple group comparisons. Spearman's rho was used to examine correlations between clinical and pathological variables. As this was intended to be an exploratory analysis, no adjustment for multiple comparisons was made.
Results
Frequency of mutations in the cohort
The C9ORF72 mutation was identified in 16/29 (55%) FTLD-TDP probands or one of their affected first-degree relatives (Table 1). All eight families in our series with autosomal dominant disease and a combination of FTD and ALS were found to have the C9ORF72 mutation. Of the 15 autosomal dominant families with clinically pure FTD, eight (53%) were explained by the C9ORF72 mutation while the other seven (47%) had a GRN mutation. None of the six cases with sporadic disease (n = 3) or a questionable family history (n = 3) was found to have mutations in either C9ORF72 or GRN; this included two cases with FTD and ALS.
The following sections describe only those families and subjects with the C9ORF72 mutation.
Families with the C9ORF72 mutation
Pedigrees of the 16 families with the C9ORF72 mutation are demonstrated in Supplementary Fig. 1. All showed a pattern of inheritance consistent with autosomal dominant disease with high penetrance. In eight families (50%) the clinical phenotype included both FTD and ALS while in the other eight (50%) the phenotype was only FTD. In five families, DNA was available from multiple affected members and all showed segregation of the mutation with clinical disease.
Subject demographics
All families with the C9ORF72 mutation were of European ethnic origin, including English, Scottish, Irish, German, Icelandic, Swedish, Dutch and Greek descent. Thirty affected members from the 16 families with the C9ORF72 mutation had sufficient clinical information for analysis. Twenty-one (70%) were male and nine female (Table 2). Twenty-seven subjects were deceased and three living subjects are currently being followed. The mutation had been confirmed in 21, while the other nine were affected relatives of proven mutation carriers.
Demographic information, clinical phenotype and disease course of study subjects
| Subject | Sex | Mutation | Initial diagnosis | Final diagnosis | FTD subtype | Dementia onset (years) | ALS onset (years) | Death (years) | Duration (years) |
|---|---|---|---|---|---|---|---|---|---|
| A-1 | M | NA | Alcoholic dementia | FTD–ALS | bvFTD | 56 | 60 | 62 | 6 |
| A-2 | M | Yes | ALS | ALS | 59 | 62 | 3 | ||
| A-3 | F | Yes | ALS | ALS | 74 | 76 | 2 | ||
| B-1 | M | Yes | bvFTD | FTD | bvFTD | 66 | 71 | 5 | |
| B-2 | M | NA | bvFTD | FTD | bvFTD | 58 | 66 | 8 | |
| C-1 | F | NA | PNFA | FTD–ALS | PNFA/bvFTD | 54 | 56 | 58 | 4 |
| C-2 | M | Yes | PNFA | FTD | PNFA/bvFTD | 67 | 76 | 9 | |
| C-3 | M | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 52 | 54 | 55 | 3 |
| C-4 | F | NA | ALS | ALS | 53 | 56 | 3 | ||
| D-1 | M | Yes | ALS | FTD–ALS | PNFA | 55 | 54 | 56 | 2 |
| D-2 | M | NA | ALS | ALS | 72 | 75 | 3 | ||
| E-1 | F | Yes | PNFA-ALS | FTD–ALS | PNFA/bvFTD | 39 | 39 | 41 | 2 |
| F-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 69 | 12 | |
| G-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 72 | 16 | |
| G-2 | M | Yes | bvFTD | FTD | bvFTD | 42 | Alive (52) | >10 | |
| H-1 | M | Yes | ALS | ALS | 45 | 47 | 2 | ||
| H-2 | M | NA | ALS | ALS | 58 | 59 | 1 | ||
| H-3 | F | NA | bvFTD | FTD | bvFTD | 40 | Alive (54) | >14 | |
| I-1 | F | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 56 | 57 | 58 | 2 |
| I-2 | M | Yes | ALS | ALS | 53 | 55 | 2 | ||
| J-1 | M | Yes | bvFTD | FTD | bvFTD | 58 | 66 | 9 | |
| K-1 | M | Yes | bvFTD | FTD–ALS | bvFTD | 50 | 53 | 56 | 6 |
| K-2 | F | NA | ALS | ALS | 61 | 62 | 1 | ||
| L-1 | M | Yes | Amnestic MCI | FTD | PNFA/bvFTD | 51 | 61 | 10 | |
| M-1 | F | Yes | bvFTD, park | FTD, park | bvFTD | 34 | 43 | 9 | |
| M-2 | M | Yes | bvFTD | FTD | bvFTD | 36 | Alive (39) | >3 | |
| M-3 | M | NA | Alcoholic dementia | FTD | bvFTD | 54 | 62 | 8 | |
| N-1 | M | Yes | Atypical AD | FTD | bvFTD | 74 | 84 | 10 | |
| O-1 | M | Yes | bvFTD | FTD | bvFTD | 53 | 56 | 3 | |
| P-1 | F | Yes | Psychosis | FTD | bvFTD | 49 | 51 | 2 | |
| mean | 53.0 ± 9.7 | 56.5 ± 8.8 | 61.3 ± 10.2 | 5.3 ± 3.9 |
| Subject | Sex | Mutation | Initial diagnosis | Final diagnosis | FTD subtype | Dementia onset (years) | ALS onset (years) | Death (years) | Duration (years) |
|---|---|---|---|---|---|---|---|---|---|
| A-1 | M | NA | Alcoholic dementia | FTD–ALS | bvFTD | 56 | 60 | 62 | 6 |
| A-2 | M | Yes | ALS | ALS | 59 | 62 | 3 | ||
| A-3 | F | Yes | ALS | ALS | 74 | 76 | 2 | ||
| B-1 | M | Yes | bvFTD | FTD | bvFTD | 66 | 71 | 5 | |
| B-2 | M | NA | bvFTD | FTD | bvFTD | 58 | 66 | 8 | |
| C-1 | F | NA | PNFA | FTD–ALS | PNFA/bvFTD | 54 | 56 | 58 | 4 |
| C-2 | M | Yes | PNFA | FTD | PNFA/bvFTD | 67 | 76 | 9 | |
| C-3 | M | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 52 | 54 | 55 | 3 |
| C-4 | F | NA | ALS | ALS | 53 | 56 | 3 | ||
| D-1 | M | Yes | ALS | FTD–ALS | PNFA | 55 | 54 | 56 | 2 |
| D-2 | M | NA | ALS | ALS | 72 | 75 | 3 | ||
| E-1 | F | Yes | PNFA-ALS | FTD–ALS | PNFA/bvFTD | 39 | 39 | 41 | 2 |
| F-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 69 | 12 | |
| G-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 72 | 16 | |
| G-2 | M | Yes | bvFTD | FTD | bvFTD | 42 | Alive (52) | >10 | |
| H-1 | M | Yes | ALS | ALS | 45 | 47 | 2 | ||
| H-2 | M | NA | ALS | ALS | 58 | 59 | 1 | ||
| H-3 | F | NA | bvFTD | FTD | bvFTD | 40 | Alive (54) | >14 | |
| I-1 | F | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 56 | 57 | 58 | 2 |
| I-2 | M | Yes | ALS | ALS | 53 | 55 | 2 | ||
| J-1 | M | Yes | bvFTD | FTD | bvFTD | 58 | 66 | 9 | |
| K-1 | M | Yes | bvFTD | FTD–ALS | bvFTD | 50 | 53 | 56 | 6 |
| K-2 | F | NA | ALS | ALS | 61 | 62 | 1 | ||
| L-1 | M | Yes | Amnestic MCI | FTD | PNFA/bvFTD | 51 | 61 | 10 | |
| M-1 | F | Yes | bvFTD, park | FTD, park | bvFTD | 34 | 43 | 9 | |
| M-2 | M | Yes | bvFTD | FTD | bvFTD | 36 | Alive (39) | >3 | |
| M-3 | M | NA | Alcoholic dementia | FTD | bvFTD | 54 | 62 | 8 | |
| N-1 | M | Yes | Atypical AD | FTD | bvFTD | 74 | 84 | 10 | |
| O-1 | M | Yes | bvFTD | FTD | bvFTD | 53 | 56 | 3 | |
| P-1 | F | Yes | Psychosis | FTD | bvFTD | 49 | 51 | 2 | |
| mean | 53.0 ± 9.7 | 56.5 ± 8.8 | 61.3 ± 10.2 | 5.3 ± 3.9 |
AD = Alzheimer's disease; bvFTD = behavioural variant frontotemporal dementia; F = female; M = male; MCI = mild cognitive impairment; NA = not available; park = parkinsonism.
Demographic information, clinical phenotype and disease course of study subjects
| Subject | Sex | Mutation | Initial diagnosis | Final diagnosis | FTD subtype | Dementia onset (years) | ALS onset (years) | Death (years) | Duration (years) |
|---|---|---|---|---|---|---|---|---|---|
| A-1 | M | NA | Alcoholic dementia | FTD–ALS | bvFTD | 56 | 60 | 62 | 6 |
| A-2 | M | Yes | ALS | ALS | 59 | 62 | 3 | ||
| A-3 | F | Yes | ALS | ALS | 74 | 76 | 2 | ||
| B-1 | M | Yes | bvFTD | FTD | bvFTD | 66 | 71 | 5 | |
| B-2 | M | NA | bvFTD | FTD | bvFTD | 58 | 66 | 8 | |
| C-1 | F | NA | PNFA | FTD–ALS | PNFA/bvFTD | 54 | 56 | 58 | 4 |
| C-2 | M | Yes | PNFA | FTD | PNFA/bvFTD | 67 | 76 | 9 | |
| C-3 | M | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 52 | 54 | 55 | 3 |
| C-4 | F | NA | ALS | ALS | 53 | 56 | 3 | ||
| D-1 | M | Yes | ALS | FTD–ALS | PNFA | 55 | 54 | 56 | 2 |
| D-2 | M | NA | ALS | ALS | 72 | 75 | 3 | ||
| E-1 | F | Yes | PNFA-ALS | FTD–ALS | PNFA/bvFTD | 39 | 39 | 41 | 2 |
| F-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 69 | 12 | |
| G-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 72 | 16 | |
| G-2 | M | Yes | bvFTD | FTD | bvFTD | 42 | Alive (52) | >10 | |
| H-1 | M | Yes | ALS | ALS | 45 | 47 | 2 | ||
| H-2 | M | NA | ALS | ALS | 58 | 59 | 1 | ||
| H-3 | F | NA | bvFTD | FTD | bvFTD | 40 | Alive (54) | >14 | |
| I-1 | F | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 56 | 57 | 58 | 2 |
| I-2 | M | Yes | ALS | ALS | 53 | 55 | 2 | ||
| J-1 | M | Yes | bvFTD | FTD | bvFTD | 58 | 66 | 9 | |
| K-1 | M | Yes | bvFTD | FTD–ALS | bvFTD | 50 | 53 | 56 | 6 |
| K-2 | F | NA | ALS | ALS | 61 | 62 | 1 | ||
| L-1 | M | Yes | Amnestic MCI | FTD | PNFA/bvFTD | 51 | 61 | 10 | |
| M-1 | F | Yes | bvFTD, park | FTD, park | bvFTD | 34 | 43 | 9 | |
| M-2 | M | Yes | bvFTD | FTD | bvFTD | 36 | Alive (39) | >3 | |
| M-3 | M | NA | Alcoholic dementia | FTD | bvFTD | 54 | 62 | 8 | |
| N-1 | M | Yes | Atypical AD | FTD | bvFTD | 74 | 84 | 10 | |
| O-1 | M | Yes | bvFTD | FTD | bvFTD | 53 | 56 | 3 | |
| P-1 | F | Yes | Psychosis | FTD | bvFTD | 49 | 51 | 2 | |
| mean | 53.0 ± 9.7 | 56.5 ± 8.8 | 61.3 ± 10.2 | 5.3 ± 3.9 |
| Subject | Sex | Mutation | Initial diagnosis | Final diagnosis | FTD subtype | Dementia onset (years) | ALS onset (years) | Death (years) | Duration (years) |
|---|---|---|---|---|---|---|---|---|---|
| A-1 | M | NA | Alcoholic dementia | FTD–ALS | bvFTD | 56 | 60 | 62 | 6 |
| A-2 | M | Yes | ALS | ALS | 59 | 62 | 3 | ||
| A-3 | F | Yes | ALS | ALS | 74 | 76 | 2 | ||
| B-1 | M | Yes | bvFTD | FTD | bvFTD | 66 | 71 | 5 | |
| B-2 | M | NA | bvFTD | FTD | bvFTD | 58 | 66 | 8 | |
| C-1 | F | NA | PNFA | FTD–ALS | PNFA/bvFTD | 54 | 56 | 58 | 4 |
| C-2 | M | Yes | PNFA | FTD | PNFA/bvFTD | 67 | 76 | 9 | |
| C-3 | M | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 52 | 54 | 55 | 3 |
| C-4 | F | NA | ALS | ALS | 53 | 56 | 3 | ||
| D-1 | M | Yes | ALS | FTD–ALS | PNFA | 55 | 54 | 56 | 2 |
| D-2 | M | NA | ALS | ALS | 72 | 75 | 3 | ||
| E-1 | F | Yes | PNFA-ALS | FTD–ALS | PNFA/bvFTD | 39 | 39 | 41 | 2 |
| F-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 69 | 12 | |
| G-1 | M | Yes | bvFTD | FTD | bvFTD | 56 | 72 | 16 | |
| G-2 | M | Yes | bvFTD | FTD | bvFTD | 42 | Alive (52) | >10 | |
| H-1 | M | Yes | ALS | ALS | 45 | 47 | 2 | ||
| H-2 | M | NA | ALS | ALS | 58 | 59 | 1 | ||
| H-3 | F | NA | bvFTD | FTD | bvFTD | 40 | Alive (54) | >14 | |
| I-1 | F | Yes | PNFA | FTD–ALS | PNFA/bvFTD | 56 | 57 | 58 | 2 |
| I-2 | M | Yes | ALS | ALS | 53 | 55 | 2 | ||
| J-1 | M | Yes | bvFTD | FTD | bvFTD | 58 | 66 | 9 | |
| K-1 | M | Yes | bvFTD | FTD–ALS | bvFTD | 50 | 53 | 56 | 6 |
| K-2 | F | NA | ALS | ALS | 61 | 62 | 1 | ||
| L-1 | M | Yes | Amnestic MCI | FTD | PNFA/bvFTD | 51 | 61 | 10 | |
| M-1 | F | Yes | bvFTD, park | FTD, park | bvFTD | 34 | 43 | 9 | |
| M-2 | M | Yes | bvFTD | FTD | bvFTD | 36 | Alive (39) | >3 | |
| M-3 | M | NA | Alcoholic dementia | FTD | bvFTD | 54 | 62 | 8 | |
| N-1 | M | Yes | Atypical AD | FTD | bvFTD | 74 | 84 | 10 | |
| O-1 | M | Yes | bvFTD | FTD | bvFTD | 53 | 56 | 3 | |
| P-1 | F | Yes | Psychosis | FTD | bvFTD | 49 | 51 | 2 | |
| mean | 53.0 ± 9.7 | 56.5 ± 8.8 | 61.3 ± 10.2 | 5.3 ± 3.9 |
AD = Alzheimer's disease; bvFTD = behavioural variant frontotemporal dementia; F = female; M = male; MCI = mild cognitive impairment; NA = not available; park = parkinsonism.
Fifteen subjects had a final clinical diagnosis of FTD, eight ALS and seven had both FTD and ALS (FTD–ALS). Of those with FTD–ALS, five began with FTD, one with ALS and one had synchronous onset. The mean age of onset of all clinical symptoms was 54.3 ± 10.2 years (range = 34–74 years), age of death was 61.5 ± 9.9 years (range = 41–84 years) and the disease duration was 5.3 ± 3.9 years (range = 1–16 years). Cognitive dysfunction tended to start slightly earlier than ALS (52.6 ± 9.6 versus 56.5 ± 8.8, not significant). The only significant difference in the disease course between the clinical groups was that those with ALS (±FTD) had a shorter duration than those with only FTD (2.8 ± 1.5 versus 8.4 ± 3.8 years, P = 0.0002). There was a trend for the age of onset in each subsequent generation to be younger (genetic anticipation), with the mean age of onset in the parents of probands at 61.3 ± 7.7 years, the probands at 53.8 ± 10.4 years and the children of the probands at 47.0 ± 7.1 years (ANOVA, P = 0.14) (Fig. 1).
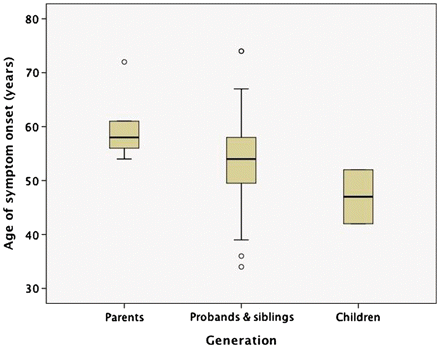
Age of onset by generation. There was a trend for the age of onset in each subsequent generation to be younger (ANOVA, P = 0.14). Boxes represent the 25–75th percentile, whiskers represent 95% confidence interval and circles are individual outliers.
Clinical features
The initial clinical diagnoses were FTD (n = 15), ALS (n = 9), FTD–ALS (n = 1), alcoholic dementia (n = 2), mild cognitive impairment (n = 1), psychosis/schizophrenia (n = 1) and atypical Alzheimer's disease (n = 1) (Table 2). The patients who were initially thought to have alcoholic dementia, atypical Alzheimer's disease and the one who presented with delusions were all subsequently diagnosed with behavioural variant FTD. No other subjects had delusions or hallucinations severe enough to fulfil a diagnosis of psychosis. One patient, who was initially diagnosed with amnestic mild cognitive amnesia because of difficulty with word recall, developed progressive non-fluent aphasia (PNFA) after 4 years.
The final clinical diagnoses were FTD (n = 15), ALS (n = 8) and FTD–ALS (n = 7). For those with a final phenotype that included FTD (n = 22), the specific subtype(s) included behavioural variant FTD (n = 15), PNFA (n = 1) and a combination of PNFA and behavioural variant FTD (n = 6). No patient was diagnosed with semantic dementia. The most common features of behavioural variant FTD included decline in personal care, disinhibition, poor judgement, apathy, perseveration and executive dysfunction. The one subject with an FTD diagnosis of pure PNFA and three of the eight subjects with only ALS also had mildly abnormal behaviour and mild executive dysfunction (Table 3). The seven subjects with a diagnosis of PNFA all had a combination of decreased verbal fluency and word finding difficulty. All of the subjects with a final diagnosis of behavioural variant FTD and one with pure ALS also had at least mild language abnormalities. Memory problems were quite common in patients with FTD (16/22, 73%), but were usually mild in severity and later in onset. Visuospatial problems and apraxia were uncommon.
Behavioural and cognitive features of study subjects
| Subject | Final diagnosis | FTD subtype | Domain | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Behaviour | Affect | Executive | Language | Memory | Visuospatial | Praxis | |||
| A-1 | FTD–ALS | bvFTD | 2 | 2 | 2 | 1 | 2 | 2 | 0 |
| A-2 | ALS | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| A-3 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| B-1 | FTD | bvFTD | 1 | 2 | 2 | 1 | 2 | 0 | 0 |
| B-2 | FTD | bvFTD | 2 | 1 | 2 | 2 | 2 | 0 | 0 |
| C-1 | FTD–ALS | PNFA/bvFTD | 2 | 2 | 2 | 3 | 1 | 0 | 0 |
| C-2 | FTD | PNFA/bvFTD | 2 | 1 | 2 | 2 | 1 | 1 | 0 |
| C-3 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 2 | 2 | 0 | 0 | 0 |
| C-4 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| D-1 | FTD–ALS | PNFA | 1 | 0 | NA | 2 | 1 | 0 | NA |
| D-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| E-1 | FTD–ALS | PNFA/bvFTD | 1 | 1 | 2 | 2 | 1 | 0 | 2 |
| F-1 | FTD | bvFTD | 3 | 2 | 2 | 1 | 1 | 1 | 1 |
| G-1 | FTD | bvFTD | 2 | NA | 2 | 1 | 1 | 2 | NA |
| G-2 | FTD | bvFTD | 2 | 1 | 1 | 1 | 0 | 1 | 0 |
| H-1 | ALS | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| H-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| H-3 | FTD | bvFTD | 2 | NA | 1 | 1 | 0 | 1 | 0 |
| I-1 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 1 | 3 | 0 | 0 | 0 |
| I-2 | ALS | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| J-1 | FTD | bvFTD | 2 | 1 | 2 | 1 | 0 | 0 | 0 |
| K-1 | FTD–ALS | bvFTD | 2 | 3 | 2 | 1 | 1 | 0 | 0 |
| K-2 | ALS | 1 | 0 | 1 | 0 | 0 | 0 | 0 | |
| L-1 | FTD | PNFA/bvFTD | 2 | 0 | 1 | 1 | 2 | 0 | 0 |
| M-1 | FTD + park | bvFTD | 1 | 3 | 2 | 2 | 1 | 1 | 1 |
| M-2 | FTD | bvFTD | 2 | 0 | 2 | 1 | 1 | 0 | 1 |
| M-3 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
| N-1 | FTD | bvFTD | 3 | 1 | 2 | 1 | 1 | 1 | 0 |
| O-1 | FTD | bvFTD | 3 | 3 | 3 | 1 | 0 | 0 | 0 |
| P-1 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
| Subject | Final diagnosis | FTD subtype | Domain | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Behaviour | Affect | Executive | Language | Memory | Visuospatial | Praxis | |||
| A-1 | FTD–ALS | bvFTD | 2 | 2 | 2 | 1 | 2 | 2 | 0 |
| A-2 | ALS | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| A-3 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| B-1 | FTD | bvFTD | 1 | 2 | 2 | 1 | 2 | 0 | 0 |
| B-2 | FTD | bvFTD | 2 | 1 | 2 | 2 | 2 | 0 | 0 |
| C-1 | FTD–ALS | PNFA/bvFTD | 2 | 2 | 2 | 3 | 1 | 0 | 0 |
| C-2 | FTD | PNFA/bvFTD | 2 | 1 | 2 | 2 | 1 | 1 | 0 |
| C-3 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 2 | 2 | 0 | 0 | 0 |
| C-4 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| D-1 | FTD–ALS | PNFA | 1 | 0 | NA | 2 | 1 | 0 | NA |
| D-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| E-1 | FTD–ALS | PNFA/bvFTD | 1 | 1 | 2 | 2 | 1 | 0 | 2 |
| F-1 | FTD | bvFTD | 3 | 2 | 2 | 1 | 1 | 1 | 1 |
| G-1 | FTD | bvFTD | 2 | NA | 2 | 1 | 1 | 2 | NA |
| G-2 | FTD | bvFTD | 2 | 1 | 1 | 1 | 0 | 1 | 0 |
| H-1 | ALS | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| H-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| H-3 | FTD | bvFTD | 2 | NA | 1 | 1 | 0 | 1 | 0 |
| I-1 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 1 | 3 | 0 | 0 | 0 |
| I-2 | ALS | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| J-1 | FTD | bvFTD | 2 | 1 | 2 | 1 | 0 | 0 | 0 |
| K-1 | FTD–ALS | bvFTD | 2 | 3 | 2 | 1 | 1 | 0 | 0 |
| K-2 | ALS | 1 | 0 | 1 | 0 | 0 | 0 | 0 | |
| L-1 | FTD | PNFA/bvFTD | 2 | 0 | 1 | 1 | 2 | 0 | 0 |
| M-1 | FTD + park | bvFTD | 1 | 3 | 2 | 2 | 1 | 1 | 1 |
| M-2 | FTD | bvFTD | 2 | 0 | 2 | 1 | 1 | 0 | 1 |
| M-3 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
| N-1 | FTD | bvFTD | 3 | 1 | 2 | 1 | 1 | 1 | 0 |
| O-1 | FTD | bvFTD | 3 | 3 | 3 | 1 | 0 | 0 | 0 |
| P-1 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
Semiquantitative grading: 0, absent; 1, mild; 2, moderate; 3, severe.
NA = not available; bvFTD = behavioural variant frontotemporal dementia; park = parkinsonism.
Behavioural and cognitive features of study subjects
| Subject | Final diagnosis | FTD subtype | Domain | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Behaviour | Affect | Executive | Language | Memory | Visuospatial | Praxis | |||
| A-1 | FTD–ALS | bvFTD | 2 | 2 | 2 | 1 | 2 | 2 | 0 |
| A-2 | ALS | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| A-3 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| B-1 | FTD | bvFTD | 1 | 2 | 2 | 1 | 2 | 0 | 0 |
| B-2 | FTD | bvFTD | 2 | 1 | 2 | 2 | 2 | 0 | 0 |
| C-1 | FTD–ALS | PNFA/bvFTD | 2 | 2 | 2 | 3 | 1 | 0 | 0 |
| C-2 | FTD | PNFA/bvFTD | 2 | 1 | 2 | 2 | 1 | 1 | 0 |
| C-3 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 2 | 2 | 0 | 0 | 0 |
| C-4 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| D-1 | FTD–ALS | PNFA | 1 | 0 | NA | 2 | 1 | 0 | NA |
| D-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| E-1 | FTD–ALS | PNFA/bvFTD | 1 | 1 | 2 | 2 | 1 | 0 | 2 |
| F-1 | FTD | bvFTD | 3 | 2 | 2 | 1 | 1 | 1 | 1 |
| G-1 | FTD | bvFTD | 2 | NA | 2 | 1 | 1 | 2 | NA |
| G-2 | FTD | bvFTD | 2 | 1 | 1 | 1 | 0 | 1 | 0 |
| H-1 | ALS | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| H-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| H-3 | FTD | bvFTD | 2 | NA | 1 | 1 | 0 | 1 | 0 |
| I-1 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 1 | 3 | 0 | 0 | 0 |
| I-2 | ALS | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| J-1 | FTD | bvFTD | 2 | 1 | 2 | 1 | 0 | 0 | 0 |
| K-1 | FTD–ALS | bvFTD | 2 | 3 | 2 | 1 | 1 | 0 | 0 |
| K-2 | ALS | 1 | 0 | 1 | 0 | 0 | 0 | 0 | |
| L-1 | FTD | PNFA/bvFTD | 2 | 0 | 1 | 1 | 2 | 0 | 0 |
| M-1 | FTD + park | bvFTD | 1 | 3 | 2 | 2 | 1 | 1 | 1 |
| M-2 | FTD | bvFTD | 2 | 0 | 2 | 1 | 1 | 0 | 1 |
| M-3 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
| N-1 | FTD | bvFTD | 3 | 1 | 2 | 1 | 1 | 1 | 0 |
| O-1 | FTD | bvFTD | 3 | 3 | 3 | 1 | 0 | 0 | 0 |
| P-1 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
| Subject | Final diagnosis | FTD subtype | Domain | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Behaviour | Affect | Executive | Language | Memory | Visuospatial | Praxis | |||
| A-1 | FTD–ALS | bvFTD | 2 | 2 | 2 | 1 | 2 | 2 | 0 |
| A-2 | ALS | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| A-3 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| B-1 | FTD | bvFTD | 1 | 2 | 2 | 1 | 2 | 0 | 0 |
| B-2 | FTD | bvFTD | 2 | 1 | 2 | 2 | 2 | 0 | 0 |
| C-1 | FTD–ALS | PNFA/bvFTD | 2 | 2 | 2 | 3 | 1 | 0 | 0 |
| C-2 | FTD | PNFA/bvFTD | 2 | 1 | 2 | 2 | 1 | 1 | 0 |
| C-3 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 2 | 2 | 0 | 0 | 0 |
| C-4 | ALS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| D-1 | FTD–ALS | PNFA | 1 | 0 | NA | 2 | 1 | 0 | NA |
| D-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| E-1 | FTD–ALS | PNFA/bvFTD | 1 | 1 | 2 | 2 | 1 | 0 | 2 |
| F-1 | FTD | bvFTD | 3 | 2 | 2 | 1 | 1 | 1 | 1 |
| G-1 | FTD | bvFTD | 2 | NA | 2 | 1 | 1 | 2 | NA |
| G-2 | FTD | bvFTD | 2 | 1 | 1 | 1 | 0 | 1 | 0 |
| H-1 | ALS | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |
| H-2 | ALS | NA | NA | NA | NA | NA | NA | NA | |
| H-3 | FTD | bvFTD | 2 | NA | 1 | 1 | 0 | 1 | 0 |
| I-1 | FTD–ALS | PNFA/bvFTD | 1 | 2 | 1 | 3 | 0 | 0 | 0 |
| I-2 | ALS | 1 | 0 | 1 | 1 | 0 | 0 | 0 | |
| J-1 | FTD | bvFTD | 2 | 1 | 2 | 1 | 0 | 0 | 0 |
| K-1 | FTD–ALS | bvFTD | 2 | 3 | 2 | 1 | 1 | 0 | 0 |
| K-2 | ALS | 1 | 0 | 1 | 0 | 0 | 0 | 0 | |
| L-1 | FTD | PNFA/bvFTD | 2 | 0 | 1 | 1 | 2 | 0 | 0 |
| M-1 | FTD + park | bvFTD | 1 | 3 | 2 | 2 | 1 | 1 | 1 |
| M-2 | FTD | bvFTD | 2 | 0 | 2 | 1 | 1 | 0 | 1 |
| M-3 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
| N-1 | FTD | bvFTD | 3 | 1 | 2 | 1 | 1 | 1 | 0 |
| O-1 | FTD | bvFTD | 3 | 3 | 3 | 1 | 0 | 0 | 0 |
| P-1 | FTD | bvFTD | 3 | 1 | 3 | 1 | 1 | 0 | 0 |
Semiquantitative grading: 0, absent; 1, mild; 2, moderate; 3, severe.
NA = not available; bvFTD = behavioural variant frontotemporal dementia; park = parkinsonism.
Fifteen subjects had a clinical diagnosis of ALS with evidence of both upper and lower motor neuron involvement (Table 4). Weakness tended to be distal and 47% of this group had prominent bulbar features. Of the subjects with a clinical diagnosis of only FTD, four (27%) had at least mild pyramidal system dysfunction, with a predominance of upper motor neuron and bulbar findings. Other motor abnormalities were common but usually mild. An extrapyramidal syndrome was present in 12 and was a prominent and presenting feature in one patient. Extrapyramidal features were usually limited to akinetic rigidity, with only four having true parkinsonism with associated resting tremor. Six patients were reported to have some combination of ataxia, unsteady gait, poor balance and abnormal position/vibration sense. Supranuclear gaze palsy was present in two patients and one had dystonic hand posturing. Urinary incontinence was a significant problem, early in the disease, for four subjects.
Motor features of study subjects
| Subject | Final diagnosis | Pattern of ALS features | Extra pyramidal | Other | |||
|---|---|---|---|---|---|---|---|
| UMN | LMN | Distal/Proximal | Bulbar | ||||
| A-1 | FTD–ALS | 1 | 2 | Distal | 0 | 2 | Unsteady gait |
| A-2 | ALS | 2 | 2 | Distal | 0 | 0 | ↓ Position/vibration sense |
| A-3 | ALS | 1 | 2 | Distal | 3 | 0 | |
| B-1 | FTD | 0 | 0 | 0 | 1 | 1 | |
| B-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| C-1 | FTD–ALS | 1 | 1 | Distal/prox | 2 | 1 | |
| C-2 | FTD | 0 | 1 | NA | NA | 0 | Incontinence |
| C-3 | FTD–ALS | 1 | 2 | Distal | 2 | 1 | |
| C-4 | ALS | 2 | 2 | Distal/prox | 1 | 0 | |
| D-1 | FTD–ALS | 2 | 2 | Distal | 1 | 1 | |
| D-2 | ALS | 2 | 1 | Distal | 2 | 0 | |
| E-1 | FTD–ALS | 2 | 2 | Distal | 1 | 2 | Gaze palsy |
| F-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| G-1 | FTD | NA | NA | NA | NA | NA | |
| G-2 | FTD | 0 | 0 | 0 | 0 | 0 | |
| H-1 | ALS | 2 | 2 | Distal | 1 | 0 | |
| H-2 | FTD | NA | NA | NA | NA | NA | |
| H-3 | ALS | NA | NA | NA | NA | NA | |
| I-1 | FTD–ALS | 2 | 1 | Distal | 2 | 0 | |
| I-2 | ALS | 2 | 1 | NA | 2 | 0 | |
| J-1 | FTD | NA | NA | NA | NA | NA | |
| K-1 | FTD–ALS | 2 | 2 | Distal | 2 | 0 | |
| K-2 | ALS | NA | NA | NA | NA | NA | |
| L-1 | FTD | 1 | 0 | 0 | 1 | 1 | ↓ Balance |
| M-1 | FTD + park | 2 | 0 | Distal/prox | 1 | 2 | ↓ Balance, dystonia, ataxia, incontinence |
| M-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| M-3 | FTD | 0 | 0 | 0 | 0 | 1 | ↓ Balance |
| N-1 | FTD | NA | NA | NA | NA | 1 | Unsteady gait |
| O-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| P-1 | FTD | 0 | 0 | 0 | 0 | 0 | Gaze palsy |
| Subject | Final diagnosis | Pattern of ALS features | Extra pyramidal | Other | |||
|---|---|---|---|---|---|---|---|
| UMN | LMN | Distal/Proximal | Bulbar | ||||
| A-1 | FTD–ALS | 1 | 2 | Distal | 0 | 2 | Unsteady gait |
| A-2 | ALS | 2 | 2 | Distal | 0 | 0 | ↓ Position/vibration sense |
| A-3 | ALS | 1 | 2 | Distal | 3 | 0 | |
| B-1 | FTD | 0 | 0 | 0 | 1 | 1 | |
| B-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| C-1 | FTD–ALS | 1 | 1 | Distal/prox | 2 | 1 | |
| C-2 | FTD | 0 | 1 | NA | NA | 0 | Incontinence |
| C-3 | FTD–ALS | 1 | 2 | Distal | 2 | 1 | |
| C-4 | ALS | 2 | 2 | Distal/prox | 1 | 0 | |
| D-1 | FTD–ALS | 2 | 2 | Distal | 1 | 1 | |
| D-2 | ALS | 2 | 1 | Distal | 2 | 0 | |
| E-1 | FTD–ALS | 2 | 2 | Distal | 1 | 2 | Gaze palsy |
| F-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| G-1 | FTD | NA | NA | NA | NA | NA | |
| G-2 | FTD | 0 | 0 | 0 | 0 | 0 | |
| H-1 | ALS | 2 | 2 | Distal | 1 | 0 | |
| H-2 | FTD | NA | NA | NA | NA | NA | |
| H-3 | ALS | NA | NA | NA | NA | NA | |
| I-1 | FTD–ALS | 2 | 1 | Distal | 2 | 0 | |
| I-2 | ALS | 2 | 1 | NA | 2 | 0 | |
| J-1 | FTD | NA | NA | NA | NA | NA | |
| K-1 | FTD–ALS | 2 | 2 | Distal | 2 | 0 | |
| K-2 | ALS | NA | NA | NA | NA | NA | |
| L-1 | FTD | 1 | 0 | 0 | 1 | 1 | ↓ Balance |
| M-1 | FTD + park | 2 | 0 | Distal/prox | 1 | 2 | ↓ Balance, dystonia, ataxia, incontinence |
| M-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| M-3 | FTD | 0 | 0 | 0 | 0 | 1 | ↓ Balance |
| N-1 | FTD | NA | NA | NA | NA | 1 | Unsteady gait |
| O-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| P-1 | FTD | 0 | 0 | 0 | 0 | 0 | Gaze palsy |
Semiquantitative grading: 0, absent; 1, mild; 2, moderate; 3, severe.
NA = not available; bvFTD = behavioural variant frontotemporal dementia; park = parkinsonism; LMN = lower motor neuron; prox = proximal; UMN = upper motor neuron; ↓ = reduced.
Motor features of study subjects
| Subject | Final diagnosis | Pattern of ALS features | Extra pyramidal | Other | |||
|---|---|---|---|---|---|---|---|
| UMN | LMN | Distal/Proximal | Bulbar | ||||
| A-1 | FTD–ALS | 1 | 2 | Distal | 0 | 2 | Unsteady gait |
| A-2 | ALS | 2 | 2 | Distal | 0 | 0 | ↓ Position/vibration sense |
| A-3 | ALS | 1 | 2 | Distal | 3 | 0 | |
| B-1 | FTD | 0 | 0 | 0 | 1 | 1 | |
| B-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| C-1 | FTD–ALS | 1 | 1 | Distal/prox | 2 | 1 | |
| C-2 | FTD | 0 | 1 | NA | NA | 0 | Incontinence |
| C-3 | FTD–ALS | 1 | 2 | Distal | 2 | 1 | |
| C-4 | ALS | 2 | 2 | Distal/prox | 1 | 0 | |
| D-1 | FTD–ALS | 2 | 2 | Distal | 1 | 1 | |
| D-2 | ALS | 2 | 1 | Distal | 2 | 0 | |
| E-1 | FTD–ALS | 2 | 2 | Distal | 1 | 2 | Gaze palsy |
| F-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| G-1 | FTD | NA | NA | NA | NA | NA | |
| G-2 | FTD | 0 | 0 | 0 | 0 | 0 | |
| H-1 | ALS | 2 | 2 | Distal | 1 | 0 | |
| H-2 | FTD | NA | NA | NA | NA | NA | |
| H-3 | ALS | NA | NA | NA | NA | NA | |
| I-1 | FTD–ALS | 2 | 1 | Distal | 2 | 0 | |
| I-2 | ALS | 2 | 1 | NA | 2 | 0 | |
| J-1 | FTD | NA | NA | NA | NA | NA | |
| K-1 | FTD–ALS | 2 | 2 | Distal | 2 | 0 | |
| K-2 | ALS | NA | NA | NA | NA | NA | |
| L-1 | FTD | 1 | 0 | 0 | 1 | 1 | ↓ Balance |
| M-1 | FTD + park | 2 | 0 | Distal/prox | 1 | 2 | ↓ Balance, dystonia, ataxia, incontinence |
| M-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| M-3 | FTD | 0 | 0 | 0 | 0 | 1 | ↓ Balance |
| N-1 | FTD | NA | NA | NA | NA | 1 | Unsteady gait |
| O-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| P-1 | FTD | 0 | 0 | 0 | 0 | 0 | Gaze palsy |
| Subject | Final diagnosis | Pattern of ALS features | Extra pyramidal | Other | |||
|---|---|---|---|---|---|---|---|
| UMN | LMN | Distal/Proximal | Bulbar | ||||
| A-1 | FTD–ALS | 1 | 2 | Distal | 0 | 2 | Unsteady gait |
| A-2 | ALS | 2 | 2 | Distal | 0 | 0 | ↓ Position/vibration sense |
| A-3 | ALS | 1 | 2 | Distal | 3 | 0 | |
| B-1 | FTD | 0 | 0 | 0 | 1 | 1 | |
| B-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| C-1 | FTD–ALS | 1 | 1 | Distal/prox | 2 | 1 | |
| C-2 | FTD | 0 | 1 | NA | NA | 0 | Incontinence |
| C-3 | FTD–ALS | 1 | 2 | Distal | 2 | 1 | |
| C-4 | ALS | 2 | 2 | Distal/prox | 1 | 0 | |
| D-1 | FTD–ALS | 2 | 2 | Distal | 1 | 1 | |
| D-2 | ALS | 2 | 1 | Distal | 2 | 0 | |
| E-1 | FTD–ALS | 2 | 2 | Distal | 1 | 2 | Gaze palsy |
| F-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| G-1 | FTD | NA | NA | NA | NA | NA | |
| G-2 | FTD | 0 | 0 | 0 | 0 | 0 | |
| H-1 | ALS | 2 | 2 | Distal | 1 | 0 | |
| H-2 | FTD | NA | NA | NA | NA | NA | |
| H-3 | ALS | NA | NA | NA | NA | NA | |
| I-1 | FTD–ALS | 2 | 1 | Distal | 2 | 0 | |
| I-2 | ALS | 2 | 1 | NA | 2 | 0 | |
| J-1 | FTD | NA | NA | NA | NA | NA | |
| K-1 | FTD–ALS | 2 | 2 | Distal | 2 | 0 | |
| K-2 | ALS | NA | NA | NA | NA | NA | |
| L-1 | FTD | 1 | 0 | 0 | 1 | 1 | ↓ Balance |
| M-1 | FTD + park | 2 | 0 | Distal/prox | 1 | 2 | ↓ Balance, dystonia, ataxia, incontinence |
| M-2 | FTD | NA | NA | NA | NA | 1 | Incontinence |
| M-3 | FTD | 0 | 0 | 0 | 0 | 1 | ↓ Balance |
| N-1 | FTD | NA | NA | NA | NA | 1 | Unsteady gait |
| O-1 | FTD | 0 | 0 | 0 | 0 | 0 | |
| P-1 | FTD | 0 | 0 | 0 | 0 | 0 | Gaze palsy |
Semiquantitative grading: 0, absent; 1, mild; 2, moderate; 3, severe.
NA = not available; bvFTD = behavioural variant frontotemporal dementia; park = parkinsonism; LMN = lower motor neuron; prox = proximal; UMN = upper motor neuron; ↓ = reduced.
There was a tendency for the clinical symptoms to accumulate and phenotypes to converge during the course of disease. The variety of signs and symptoms experienced by each subject became more complex on subsequent assessments (Table 5). Whereas only one subject had features diagnostic of both FTD and ALS at the time of initial assessment, this rose to n = 7 by the late stages of their disease (Fig. 2). All six patients with an initial FTD phenotype of PNFA later developed significant abnormalities of behaviour, affect and/or executive function.
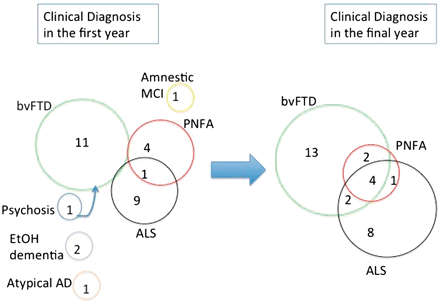
Evolution of clinical diagnoses. There was a tendency for the clinical phenotypes to converge with disease progression. AD = Alzheimer's disease; bvFTD = behavioural variant frontotemporal dementia; EtOH = alcohol related; MCI = mild cognitive impairment.
Evolution of clinical features of study subjects
| Domain Symptoms | n (%) affected in first year | n (%) affected in final year |
|---|---|---|
| Behaviour | 14 (47) | 25 (83) |
| Disinhibition | 12 (40) | 21 (70) |
| Decline in self-care | 6 (20) | 22 (73) |
| Rigidity/perseveration | 8 (27) | 15 (50) |
| Affect | 12 (40) | 20 (67) |
| Apathy | 9 (30) | 14 (47) |
| Depression | 4 (13) | 7 (23) |
| Executive | 12 (40) | 24 (80) |
| Planning/set shifting | 12 (40) | 22 (73) |
| Impaired abstraction | 10 (33) | 17 (57) |
| Poor judgement | 8 (27) | 21 (70) |
| Poor attention | 6 (20) | 12 (40) |
| Language | 14 (47) | 23 (77) |
| Reduced fluency | 12 (40) | 21 (70) |
| Word finding difficulty | 10 (33) | 17 (57) |
| Fluent aphasia | 0 (0) | 0 (0) |
| Memory (short-term) | 9 (30) | 18 (60) |
| Visuospatial | 3 (10) | 8 (27) |
| Apraxia | 1 (3) | 4 (13) |
| Extrapyramidal signs | 3 (10) | 12 (40) |
| Rigidity/bradykinesia | 2 (7) | 9 (30) |
| Tremor | 1 (3) | 4 (13) |
| ALS features | 10 (33) | 15 (50) |
| Upper motor neuron dysfunction | 7 (23) | 15 (50) |
| Lower motor neuron dysfunction | 5 (17) | 14 (47) |
| Bulbar dysfunction | 2 (7) | 14 (47) |
| Domain Symptoms | n (%) affected in first year | n (%) affected in final year |
|---|---|---|
| Behaviour | 14 (47) | 25 (83) |
| Disinhibition | 12 (40) | 21 (70) |
| Decline in self-care | 6 (20) | 22 (73) |
| Rigidity/perseveration | 8 (27) | 15 (50) |
| Affect | 12 (40) | 20 (67) |
| Apathy | 9 (30) | 14 (47) |
| Depression | 4 (13) | 7 (23) |
| Executive | 12 (40) | 24 (80) |
| Planning/set shifting | 12 (40) | 22 (73) |
| Impaired abstraction | 10 (33) | 17 (57) |
| Poor judgement | 8 (27) | 21 (70) |
| Poor attention | 6 (20) | 12 (40) |
| Language | 14 (47) | 23 (77) |
| Reduced fluency | 12 (40) | 21 (70) |
| Word finding difficulty | 10 (33) | 17 (57) |
| Fluent aphasia | 0 (0) | 0 (0) |
| Memory (short-term) | 9 (30) | 18 (60) |
| Visuospatial | 3 (10) | 8 (27) |
| Apraxia | 1 (3) | 4 (13) |
| Extrapyramidal signs | 3 (10) | 12 (40) |
| Rigidity/bradykinesia | 2 (7) | 9 (30) |
| Tremor | 1 (3) | 4 (13) |
| ALS features | 10 (33) | 15 (50) |
| Upper motor neuron dysfunction | 7 (23) | 15 (50) |
| Lower motor neuron dysfunction | 5 (17) | 14 (47) |
| Bulbar dysfunction | 2 (7) | 14 (47) |
Evolution of clinical features of study subjects
| Domain Symptoms | n (%) affected in first year | n (%) affected in final year |
|---|---|---|
| Behaviour | 14 (47) | 25 (83) |
| Disinhibition | 12 (40) | 21 (70) |
| Decline in self-care | 6 (20) | 22 (73) |
| Rigidity/perseveration | 8 (27) | 15 (50) |
| Affect | 12 (40) | 20 (67) |
| Apathy | 9 (30) | 14 (47) |
| Depression | 4 (13) | 7 (23) |
| Executive | 12 (40) | 24 (80) |
| Planning/set shifting | 12 (40) | 22 (73) |
| Impaired abstraction | 10 (33) | 17 (57) |
| Poor judgement | 8 (27) | 21 (70) |
| Poor attention | 6 (20) | 12 (40) |
| Language | 14 (47) | 23 (77) |
| Reduced fluency | 12 (40) | 21 (70) |
| Word finding difficulty | 10 (33) | 17 (57) |
| Fluent aphasia | 0 (0) | 0 (0) |
| Memory (short-term) | 9 (30) | 18 (60) |
| Visuospatial | 3 (10) | 8 (27) |
| Apraxia | 1 (3) | 4 (13) |
| Extrapyramidal signs | 3 (10) | 12 (40) |
| Rigidity/bradykinesia | 2 (7) | 9 (30) |
| Tremor | 1 (3) | 4 (13) |
| ALS features | 10 (33) | 15 (50) |
| Upper motor neuron dysfunction | 7 (23) | 15 (50) |
| Lower motor neuron dysfunction | 5 (17) | 14 (47) |
| Bulbar dysfunction | 2 (7) | 14 (47) |
| Domain Symptoms | n (%) affected in first year | n (%) affected in final year |
|---|---|---|
| Behaviour | 14 (47) | 25 (83) |
| Disinhibition | 12 (40) | 21 (70) |
| Decline in self-care | 6 (20) | 22 (73) |
| Rigidity/perseveration | 8 (27) | 15 (50) |
| Affect | 12 (40) | 20 (67) |
| Apathy | 9 (30) | 14 (47) |
| Depression | 4 (13) | 7 (23) |
| Executive | 12 (40) | 24 (80) |
| Planning/set shifting | 12 (40) | 22 (73) |
| Impaired abstraction | 10 (33) | 17 (57) |
| Poor judgement | 8 (27) | 21 (70) |
| Poor attention | 6 (20) | 12 (40) |
| Language | 14 (47) | 23 (77) |
| Reduced fluency | 12 (40) | 21 (70) |
| Word finding difficulty | 10 (33) | 17 (57) |
| Fluent aphasia | 0 (0) | 0 (0) |
| Memory (short-term) | 9 (30) | 18 (60) |
| Visuospatial | 3 (10) | 8 (27) |
| Apraxia | 1 (3) | 4 (13) |
| Extrapyramidal signs | 3 (10) | 12 (40) |
| Rigidity/bradykinesia | 2 (7) | 9 (30) |
| Tremor | 1 (3) | 4 (13) |
| ALS features | 10 (33) | 15 (50) |
| Upper motor neuron dysfunction | 7 (23) | 15 (50) |
| Lower motor neuron dysfunction | 5 (17) | 14 (47) |
| Bulbar dysfunction | 2 (7) | 14 (47) |
Clinical variation within C9ORF72 families
There was significant clinical heterogeneity, not only between families, but also within families (Table 2). For nine families, there was clinical information available for multiple affected members. The age of onset within families varied by as much as 22 years (Family M) and the duration by >14 years (Family H). In only three families did members have a consistent clinical phenotype (behavioural variant FTD); whereas, in the other six, the presentation and final diagnoses included ALS, FTD or both and the FTD could be either behavioural variant FTD or PNFA. An example of such intrafamilial variation is provided below.
Illustrative cases
The following case descriptions are included to illustrate the degree of clinical variation that was present within some individual families. The four subjects described were all related. The family designation, the relationship among the members and their exact ages, have been removed in order to protect the identity of the family.
Family member 1
This female began experiencing cognitive difficulties in her mid-fifties, with increasing forgetfulness, hesitant speech and word finding problems, resulting in the loss of her employment. Her family became concerned about her inattentiveness and difficulties with banking. On initial assessment, she was pleasant and well-groomed but giggled inappropriately. Her speech was interrupted by frequent stuttering, slurring, perseverative phrases and semantic and phonemic paraphasic errors. Mini-Mental State Examination score was 20/30 with points lost on orientation, object recall, mental reversal and sentence repetition. Her clock-drawing was mildly impaired. Neuropsychological testing identified severe impairment in letter fluency and abstraction of similarities. She had moderate impairment in visual and auditory receptive language and verbal memory acquisition and retention. Her non-verbal memory was mildly impaired. Initial neurological examination was normal apart from diffusely brisk limb reflexes. Neuroimaging demonstrated mild generalized atrophy on head CT and frontal and perisylvian hypometabolism on FDG-PET. Her initial diagnosis was PNFA because language difficulty was felt to be the most prominent deficit during the initial stage of her disease. Over the following year she progressed rapidly, becoming mute and apathetic. She developed progressive motor impairment with dysphagia, spasticity, mild rigidity and an extensor plantar response. Her EMG revealed diffuse denervation and her diagnosis was changed to FTD–ALS. She died after an illness of 4 years from respiratory complications.
Family member 2
This well-educated male noted difficulty completing his income tax return in his late sixties. In the following year, he developed progressive memory problems and changes in behaviour that included making inappropriate comments to several children. When he was initially assessed, his Mini-Mental State Examination was 22/30 with problems noted in his orientation, mental control and naming, with relative preservation of short term recall. He had difficulty interpreting proverbs and on language testing demonstrated reduced verbal fluency, significant anomia, alexia and verbal apraxia. His clock drawing was mildly impaired. The remainder of his neurological examination was normal, except for frontal release signs with positive palmomental and grasp reflexes. An initial diagnosis of PNFA was made, based on the relative severity of his language deficits. His behaviour declined progressively over the next year, with frequent agitation and aggressive outbursts. He was placed in a nursing home where he stabilized until the final year of his life when he was noted to have frequent falls. He died a decade after the onset of symptoms from respiratory complications.
Family member 3
This patient was assessed in his mid-fifties, following 2 years of progressive speech difficulty. His fluency had declined with frequent syntactical errors, and he was slow to respond in conversation. His verbal comprehension and reading abilities were described as being intact. His family had also noted more recent behavioural changes with apathy, impulsivity and poor financial planning. He had limited insight into these difficulties and denied any problems. When assessed initially he was co-operative but noted to have blunted affect. His speech was hesitant and slow, with occasional stuttering. He scored 29/30 on Mini-Mental State Examination and 93/100 on the modified Mini-Mental State Examination with points lost on verbal fluency and verbal abstract reasoning. His clock drawing was normal. On the Montreal Cognitive Assessment, he scored 24/30 with problems in naming, delayed recall, phonemic verbal fluency and sentence repetition. On the Frontal Assessment Battery, he scored 16/18 with points lost for phonemic fluency and inhibitory control. His general neurological examination was unremarkable. Neuropsychological testing demonstrated moderate impairment in language, attention and executive function with relative sparing of verbal and non-verbal memory. Neuroimaging with MRI showed frontal and temporal atrophy, greater on the left (Fig. 3A), and FDG-PET demonstrated frontal hypometabolism (Fig. 3B). He was diagnosed as having FTD with PNFA and some behavioural symptoms. Within the next year, he developed mild cognitive decline and significant new left-sided weakness, progressive slurring of speech and dysphagia. He had mixed upper and lower motor neuron signs with bilateral facial weakness, hoarseness, brisk jaw jerk and spasticity in all limbs. He had tongue atrophy with fasciculations and weakness in his neck flexors and extensors (4/5) as well as his left arm and leg (4+/5). Fasciculations were noted in his thighs. EMG study confirmed widespread denervation and his diagnosis was changed to FTD–ALS. He was treated with riluzole. Over the next 3 months, his dysphagia worsened and a percutaneous endoscopic gastrostomy tube was inserted because of aspiration risk. He died <2 years after disease onset from sepsis.
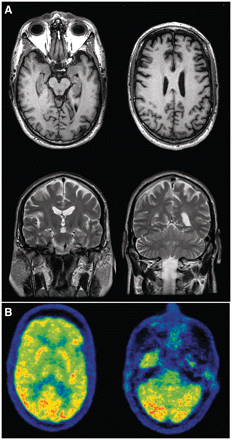
Neuroimaging on illustrative Family member 3. MRI at the age of 54 (A). Top: T1 axial images at the level of hippocampus (left) and corona radiata (right), demonstrating anterior frontal and temporal lobar atrophy (left > right). Bottom: T2 coronal images at the level of the hippocampus (left) and cerebellum (right), further demonstrating the asymmetric atrophy and showing hippocampal atrophy that is mild compared to the degree of cortical involvement. FDG-PET at the age of 54 years (B) shows hypometabolism in the frontal lobes relative to the occipital cortex at the level of the basal ganglia (left) and the cerebellum (right).
Family member 4
This patient initially presented in her early fifties with gradually progressive leg weakness. On her assessment, she was noted to have marked hyper-reflexia in all limbs and bilateral extensor plantar responses. Her MRI showed narrowing of her spinal canal at C3/C4 with possible spinal cord compression. She had decompressive spinal surgery for cervical myelopathy with initial improvement in her leg strength. However, within a few years, she developed recurrent weakness of her legs as well as her right arm. There were no obvious cognitive problems, although relatives felt she was more irritable. Neurological examination at that time revealed motor abnormalities with slow tongue movements and weakness in her right arm (4+/5) and both legs (4−/5). Muscle wasting and fasciculations were noted in her right biceps and left quadriceps. A biopsy of her right biceps showed changes of chronic denervation atrophy, consistent with ALS. Over the next 2 years, she developed progressive neck and bulbar weakness with increasing dysphagia. She died of respiratory failure after a 5-year illness.
Neuroimaging
Neuroimaging reports were available for 21 subjects, including; CT (n = 16), MRI (n = 11), FDG-PET (n = 5) and SPECT (n = 5). A summary of imaging findings is shown in Supplementary Table 1. In subjects with FTD, focal atrophy was more often detected with MRI (7/9 focal atrophy, 2/9 diffuse atrophy) compared to CT (5/15 focal atrophy, 7/15 diffuse atrophy and 3/15 normal). PET (4/5) and SPECT (3/5) helped to further identify frontal abnormalities in several subjects with non-focal structural imaging. Left–right asymmetry was reported in only one case of PNFA (Case C-3). In the three subjects who had ALS at initial presentation, one had a normal MRI, one had generalized atrophy, and one demonstrated abnormal corticospinal tract T2 signal.
Neuropathology
Autopsy material was available from 21 affected members from the 16 families. Their final clinical diagnoses were FTD (n = 11), FTD–ALS (n = 8) and ALS (n = 2). The presence of the C9ORF72 mutation had been confirmed in 18 while the other three had an affected first-degree relative with proven mutation.
The weight of the post-mortem brain specimen (mean = 1238 ± 212 g, range 720–1680 g) and the degree of cerebral atrophy varied considerably (Table 6). Gross atrophy of the cerebral lobes was noted in 14 (67%) cases, was generally symmetric and always involved the frontal lobes, with the temporal lobes less commonly affected. There was reduced pigmentation of the substantia nigra in 12 (57%) cases and atrophy of the head of the caudate nucleus in four (19%). Non-specific changes of chronic degeneration in affected regions of the cerebral cortex included neuronal loss, astrocytic gliosis, microglial activation and laminar superficial spongiosis. Chronic degenerative changes were also common in the striatum and substantia nigra. Thirteen cases showed selective pyramidal cell loss from the CA1 region of the hippocampus, which was usually mild or moderate. There was reduced myelin staining of the corticospinal tracts in 14 cases and loss of lower motor neurons in all but one of the cases where the spinal cord was available. Other brain regions were inconsistently affected or spared.
Neuropathological findings of study subjects
| Case | Final clinical diagnosis | Brain weight (g) | Gross atrophy | Microscopic changes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL | TL | BG | SN | FC | TC | NC | HC CA1 | HC dent | BG | SN | CST | LMN | cer ctx ub | FTLD-TDP | Other | |||||||
| deg | deg | TDP | deg | TDP | TDP | deg | TDP | deg | TDP | deg | deg | TDP | type | |||||||||
| A-1 | FTD–ALS | 1680 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 1 | 2 | 2 | 2 | 0 | 1 | 2 | 3 | 3 | 3 | B + A | |
| A-3 | ALS | 1296 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 1 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | B | |
| B-1 | FTD | 1230 | 1 | 1 | 0 | 1 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 2 | 1 | 2 | 1 | 1a | 2 a | 3 | B + A | |
| B-2 | FTD | 1480 | 2 | 3 | 0 | 0 | 2 | 2 | 3 | 1 | 3 | 3 | 1 | 2 | 2 | 1 | NA | NA | NA | 3 | B + A | |
| C-1 | FTD–ALS | 1400 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 0 | 3 | 1 | 2 | NA | 2 | NA | NA | NA | NA | B | |
| C-2 | FTD | 1150 | 2 | 2 | 0 | 1 | 1 | 2 | 1 | 1 | 3 | 3 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | B | AD |
| C-3 | FTD–ALS | NA | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 3 | 1 | 2 | 2 | 3 | 3 | 2 | B | |
| D-1 | FTD–ALS | 1470 | 1 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 1 | 2 | 3 | NA | B | |
| E-1 | FTD–ALS | 970 | 2 | 0 | 1 | 1 | 3 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| E-2 | FTD–ALS | 1130 | 2 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | 3 | NA | NA | NA | NA | 2 | 1 a | 2 a | 1 | B | |
| F-1 | FTD | 1300 | 2 | 2 | 3 | 2 | 2 | 2 | 3 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | 1 | 1 | 1 | 3 | B + A | AD |
| G-1 | FTD | 1165 | 0 | 0 | 0 | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 2 | 3 | 2 | 2 | 1 | 0 a | 1 a | 3 | B + A | AD |
| H-1 | ALS | NA | 0 | 0 | 0 | 0 | 1 | NA | 2 | 1 | 0 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| I-1 | FTD–ALS | 1175 | 2 | 1 | 1 | 1 | 2 | 2 | 3 | 2 | 0 | 3 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 2 | B | |
| J-1 | FTD | 1400 | 1 | 2 | 0 | 1 | 1 | 2 | 2 | 2 | 2 | 3 | 1 | 1 | 2 | 1 | 0 | 1 | 2 | 2 | B | |
| K-1 | FTD–ALS | NA | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 3 | B | |
| L-1 | FTD | 1242 | 3 | 0 | 0 | 2 | 2 | 0 | 3 | 0 | 1 | 3 | 1 | 3 | 2 | 2 | 0 | 1 | 1 | 2 | B + A | |
| M-1 | FTD + park | 720 | 3 | 3 | 3 | 0 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 1 | 1 | 3 | B + A | |
| N-1 | FTD | 1216 | 0 | 0 | 0 | 0 | 1 | NA | 2 | 3 | 2 | 3 | 2 | NA | 1 | 1 | 1 | 0 a | 1 a | NA | B + A | |
| O-1 | FTD | 1075 | 2 | 2 | 0 | 3 | 2 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 0 | 1 | 2 | 3 | B | |
| P-1 | FTD | 1180 | 2 | 1 | 0 | 2 | 2 | 2 | 3 | 0 | 1 | 3 | 1 | 2 | 1 | 2 | 0 | 1 | 2 | 1 | B | |
| Case | Final clinical diagnosis | Brain weight (g) | Gross atrophy | Microscopic changes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL | TL | BG | SN | FC | TC | NC | HC CA1 | HC dent | BG | SN | CST | LMN | cer ctx ub | FTLD-TDP | Other | |||||||
| deg | deg | TDP | deg | TDP | TDP | deg | TDP | deg | TDP | deg | deg | TDP | type | |||||||||
| A-1 | FTD–ALS | 1680 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 1 | 2 | 2 | 2 | 0 | 1 | 2 | 3 | 3 | 3 | B + A | |
| A-3 | ALS | 1296 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 1 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | B | |
| B-1 | FTD | 1230 | 1 | 1 | 0 | 1 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 2 | 1 | 2 | 1 | 1a | 2 a | 3 | B + A | |
| B-2 | FTD | 1480 | 2 | 3 | 0 | 0 | 2 | 2 | 3 | 1 | 3 | 3 | 1 | 2 | 2 | 1 | NA | NA | NA | 3 | B + A | |
| C-1 | FTD–ALS | 1400 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 0 | 3 | 1 | 2 | NA | 2 | NA | NA | NA | NA | B | |
| C-2 | FTD | 1150 | 2 | 2 | 0 | 1 | 1 | 2 | 1 | 1 | 3 | 3 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | B | AD |
| C-3 | FTD–ALS | NA | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 3 | 1 | 2 | 2 | 3 | 3 | 2 | B | |
| D-1 | FTD–ALS | 1470 | 1 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 1 | 2 | 3 | NA | B | |
| E-1 | FTD–ALS | 970 | 2 | 0 | 1 | 1 | 3 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| E-2 | FTD–ALS | 1130 | 2 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | 3 | NA | NA | NA | NA | 2 | 1 a | 2 a | 1 | B | |
| F-1 | FTD | 1300 | 2 | 2 | 3 | 2 | 2 | 2 | 3 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | 1 | 1 | 1 | 3 | B + A | AD |
| G-1 | FTD | 1165 | 0 | 0 | 0 | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 2 | 3 | 2 | 2 | 1 | 0 a | 1 a | 3 | B + A | AD |
| H-1 | ALS | NA | 0 | 0 | 0 | 0 | 1 | NA | 2 | 1 | 0 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| I-1 | FTD–ALS | 1175 | 2 | 1 | 1 | 1 | 2 | 2 | 3 | 2 | 0 | 3 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 2 | B | |
| J-1 | FTD | 1400 | 1 | 2 | 0 | 1 | 1 | 2 | 2 | 2 | 2 | 3 | 1 | 1 | 2 | 1 | 0 | 1 | 2 | 2 | B | |
| K-1 | FTD–ALS | NA | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 3 | B | |
| L-1 | FTD | 1242 | 3 | 0 | 0 | 2 | 2 | 0 | 3 | 0 | 1 | 3 | 1 | 3 | 2 | 2 | 0 | 1 | 1 | 2 | B + A | |
| M-1 | FTD + park | 720 | 3 | 3 | 3 | 0 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 1 | 1 | 3 | B + A | |
| N-1 | FTD | 1216 | 0 | 0 | 0 | 0 | 1 | NA | 2 | 3 | 2 | 3 | 2 | NA | 1 | 1 | 1 | 0 a | 1 a | NA | B + A | |
| O-1 | FTD | 1075 | 2 | 2 | 0 | 3 | 2 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 0 | 1 | 2 | 3 | B | |
| P-1 | FTD | 1180 | 2 | 1 | 0 | 2 | 2 | 2 | 3 | 0 | 1 | 3 | 1 | 2 | 1 | 2 | 0 | 1 | 2 | 1 | B | |
a Only medulla, no spinal cord available. Semi-quantitative grading: 0, absent; 1, mild; 2, moderate; 3, severe.
NA = not available; AD = Alzheimer's disease pathology; BG = basal ganglia; CA1 = cornu ammonis zone 1; cer ctx = cerebellar cortex; CST = corticospinal tracts; deg = degenerative changes; dent = dentate granule cell layer; FC = frontal cortex; FL = frontal lobe; HC = hippocampus; LMN = lower motor nuclei (hypoglossal nucleus and spinal cord ventral grey matter); park = parkinsonism; SN = substantia nigra; TC = temporal cortex; TL = temporal lobe; ub = ubiquitin immunoreactive pathology.
Neuropathological findings of study subjects
| Case | Final clinical diagnosis | Brain weight (g) | Gross atrophy | Microscopic changes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL | TL | BG | SN | FC | TC | NC | HC CA1 | HC dent | BG | SN | CST | LMN | cer ctx ub | FTLD-TDP | Other | |||||||
| deg | deg | TDP | deg | TDP | TDP | deg | TDP | deg | TDP | deg | deg | TDP | type | |||||||||
| A-1 | FTD–ALS | 1680 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 1 | 2 | 2 | 2 | 0 | 1 | 2 | 3 | 3 | 3 | B + A | |
| A-3 | ALS | 1296 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 1 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | B | |
| B-1 | FTD | 1230 | 1 | 1 | 0 | 1 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 2 | 1 | 2 | 1 | 1a | 2 a | 3 | B + A | |
| B-2 | FTD | 1480 | 2 | 3 | 0 | 0 | 2 | 2 | 3 | 1 | 3 | 3 | 1 | 2 | 2 | 1 | NA | NA | NA | 3 | B + A | |
| C-1 | FTD–ALS | 1400 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 0 | 3 | 1 | 2 | NA | 2 | NA | NA | NA | NA | B | |
| C-2 | FTD | 1150 | 2 | 2 | 0 | 1 | 1 | 2 | 1 | 1 | 3 | 3 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | B | AD |
| C-3 | FTD–ALS | NA | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 3 | 1 | 2 | 2 | 3 | 3 | 2 | B | |
| D-1 | FTD–ALS | 1470 | 1 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 1 | 2 | 3 | NA | B | |
| E-1 | FTD–ALS | 970 | 2 | 0 | 1 | 1 | 3 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| E-2 | FTD–ALS | 1130 | 2 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | 3 | NA | NA | NA | NA | 2 | 1 a | 2 a | 1 | B | |
| F-1 | FTD | 1300 | 2 | 2 | 3 | 2 | 2 | 2 | 3 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | 1 | 1 | 1 | 3 | B + A | AD |
| G-1 | FTD | 1165 | 0 | 0 | 0 | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 2 | 3 | 2 | 2 | 1 | 0 a | 1 a | 3 | B + A | AD |
| H-1 | ALS | NA | 0 | 0 | 0 | 0 | 1 | NA | 2 | 1 | 0 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| I-1 | FTD–ALS | 1175 | 2 | 1 | 1 | 1 | 2 | 2 | 3 | 2 | 0 | 3 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 2 | B | |
| J-1 | FTD | 1400 | 1 | 2 | 0 | 1 | 1 | 2 | 2 | 2 | 2 | 3 | 1 | 1 | 2 | 1 | 0 | 1 | 2 | 2 | B | |
| K-1 | FTD–ALS | NA | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 3 | B | |
| L-1 | FTD | 1242 | 3 | 0 | 0 | 2 | 2 | 0 | 3 | 0 | 1 | 3 | 1 | 3 | 2 | 2 | 0 | 1 | 1 | 2 | B + A | |
| M-1 | FTD + park | 720 | 3 | 3 | 3 | 0 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 1 | 1 | 3 | B + A | |
| N-1 | FTD | 1216 | 0 | 0 | 0 | 0 | 1 | NA | 2 | 3 | 2 | 3 | 2 | NA | 1 | 1 | 1 | 0 a | 1 a | NA | B + A | |
| O-1 | FTD | 1075 | 2 | 2 | 0 | 3 | 2 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 0 | 1 | 2 | 3 | B | |
| P-1 | FTD | 1180 | 2 | 1 | 0 | 2 | 2 | 2 | 3 | 0 | 1 | 3 | 1 | 2 | 1 | 2 | 0 | 1 | 2 | 1 | B | |
| Case | Final clinical diagnosis | Brain weight (g) | Gross atrophy | Microscopic changes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL | TL | BG | SN | FC | TC | NC | HC CA1 | HC dent | BG | SN | CST | LMN | cer ctx ub | FTLD-TDP | Other | |||||||
| deg | deg | TDP | deg | TDP | TDP | deg | TDP | deg | TDP | deg | deg | TDP | type | |||||||||
| A-1 | FTD–ALS | 1680 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 1 | 2 | 2 | 2 | 0 | 1 | 2 | 3 | 3 | 3 | B + A | |
| A-3 | ALS | 1296 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 1 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | B | |
| B-1 | FTD | 1230 | 1 | 1 | 0 | 1 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 2 | 1 | 2 | 1 | 1a | 2 a | 3 | B + A | |
| B-2 | FTD | 1480 | 2 | 3 | 0 | 0 | 2 | 2 | 3 | 1 | 3 | 3 | 1 | 2 | 2 | 1 | NA | NA | NA | 3 | B + A | |
| C-1 | FTD–ALS | 1400 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 0 | 3 | 1 | 2 | NA | 2 | NA | NA | NA | NA | B | |
| C-2 | FTD | 1150 | 2 | 2 | 0 | 1 | 1 | 2 | 1 | 1 | 3 | 3 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | B | AD |
| C-3 | FTD–ALS | NA | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 3 | 1 | 2 | 2 | 3 | 3 | 2 | B | |
| D-1 | FTD–ALS | 1470 | 1 | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 1 | 2 | 3 | NA | B | |
| E-1 | FTD–ALS | 970 | 2 | 0 | 1 | 1 | 3 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| E-2 | FTD–ALS | 1130 | 2 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | 3 | NA | NA | NA | NA | 2 | 1 a | 2 a | 1 | B | |
| F-1 | FTD | 1300 | 2 | 2 | 3 | 2 | 2 | 2 | 3 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | 1 | 1 | 1 | 3 | B + A | AD |
| G-1 | FTD | 1165 | 0 | 0 | 0 | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 2 | 3 | 2 | 2 | 1 | 0 a | 1 a | 3 | B + A | AD |
| H-1 | ALS | NA | 0 | 0 | 0 | 0 | 1 | NA | 2 | 1 | 0 | 3 | 1 | 2 | 1 | 2 | 3 | 3 | 3 | 3 | B | |
| I-1 | FTD–ALS | 1175 | 2 | 1 | 1 | 1 | 2 | 2 | 3 | 2 | 0 | 3 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 2 | B | |
| J-1 | FTD | 1400 | 1 | 2 | 0 | 1 | 1 | 2 | 2 | 2 | 2 | 3 | 1 | 1 | 2 | 1 | 0 | 1 | 2 | 2 | B | |
| K-1 | FTD–ALS | NA | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 1 | 2 | 3 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 3 | B | |
| L-1 | FTD | 1242 | 3 | 0 | 0 | 2 | 2 | 0 | 3 | 0 | 1 | 3 | 1 | 3 | 2 | 2 | 0 | 1 | 1 | 2 | B + A | |
| M-1 | FTD + park | 720 | 3 | 3 | 3 | 0 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 1 | 1 | 3 | B + A | |
| N-1 | FTD | 1216 | 0 | 0 | 0 | 0 | 1 | NA | 2 | 3 | 2 | 3 | 2 | NA | 1 | 1 | 1 | 0 a | 1 a | NA | B + A | |
| O-1 | FTD | 1075 | 2 | 2 | 0 | 3 | 2 | 1 | 2 | 0 | 0 | 3 | 0 | 1 | 2 | 2 | 0 | 1 | 2 | 3 | B | |
| P-1 | FTD | 1180 | 2 | 1 | 0 | 2 | 2 | 2 | 3 | 0 | 1 | 3 | 1 | 2 | 1 | 2 | 0 | 1 | 2 | 1 | B | |
a Only medulla, no spinal cord available. Semi-quantitative grading: 0, absent; 1, mild; 2, moderate; 3, severe.
NA = not available; AD = Alzheimer's disease pathology; BG = basal ganglia; CA1 = cornu ammonis zone 1; cer ctx = cerebellar cortex; CST = corticospinal tracts; deg = degenerative changes; dent = dentate granule cell layer; FC = frontal cortex; FL = frontal lobe; HC = hippocampus; LMN = lower motor nuclei (hypoglossal nucleus and spinal cord ventral grey matter); park = parkinsonism; SN = substantia nigra; TC = temporal cortex; TL = temporal lobe; ub = ubiquitin immunoreactive pathology.
The microscopic neuropathology was characterized by the presence of TDP-43 immunoreactive cellular inclusions, in a wide range of neuroanatomical regions (Table 6). The cerebral neocortical pathology could be divided into two broad groups. In 13 cases (62%), the pattern was consistent with FTLD-TDP type B (equivalent to Mackenzie type 3 and Sampathu type 2) (Mackenzie et al., 2011), characterized by compact neuronal cytoplasmic inclusions in all cortical layers with relatively few dystrophic neurites (Fig. 4A). In addition, many small layer II neurons had diffuse granular cytoplasmic TDP-43 reactivity (pre-inclusions) (Fig. 4B). The remaining eight cases (38%) also showed FTLD-TDP type B features, but in combination with a superficial band of more compact small neuronal cytoplasmic inclusions and dystrophic neurites, as well as rare lentiform neuronal intranuclear inclusions (NII); a pattern consistent with FTLD-TDP type A (equivalent to Mackenzie type 1 and Sampathu type 3) (Fig. 4C) (Mackenzie et al., 2011). In the hippocampus, all cases had numerous compact and granular TDP-43 immunoreactive neuronal cytoplasmic inclusions in dentate granule cells and a majority had small thread-like processes concentrated in the CA1 region (Fig. 4D). Variable numbers of TDP-43 immunoreactive neuronal cytoplasmic inclusions and dystrophic neurites were present in many subcortical regions with the striatum and substantia nigra consistently involved. Small numbers of TDP-43 immunoreactive glial cytoplasmic inclusions were a common finding in subcortical nuclei and white matter. In all but one case (Case C-2, clinical FTD), at least some TDP-43 immunoreactive neuronal and glial inclusions were present in the ventral grey matter of the spinal cord (when available) and/or brainstem motor nuclei. The morphology of neuronal cytoplasmic inclusions in lower motor neurons included bundles of filaments, compact round bodies and diffuse cytoplasmic granules (Fig. 4E and F).
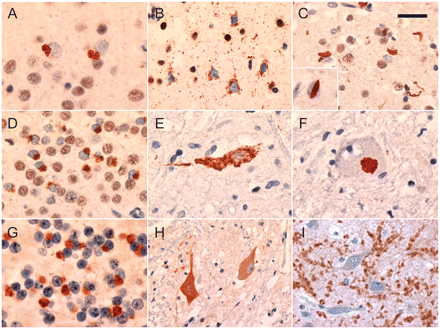
Neuropathological features associated with the C9ORF72 mutation. All cases showed compact and granular TDP-43-immunoreactive neuronal cytoplasmic inclusions in the neocortex, typical of FTLD-TDP type B (A). Granular neuronal pre-inclusions in neocortex layer II were common (B). A subset of cases had compact neuronal cytoplasmic inclusions, short neurites and rare lentiform neuronal intranuclear inclusions (inset) in layer II neocortex, consistent with FTLD-TDP type A (C). Compact and granular neuronal cytoplasmic inclusions in hippocampal dentate granule cells were a consistent feature (D). Lower motor neurons contained neuronal cytoplasmic inclusions with granular, filamentous (E) or compact Lewy body-like morphology (F). Small neuronal cytoplasmic inclusions and short neurites in the granule cell layer of the cerebellar cortex were immunoreactive for ubiquitin and p62, but negative for TDP-43 (G). Increased cytoplasmic staining of some lower motor neurons was seen in cases of ALS, both with and without the C9ORF72 mutation (H). In all cases of FTLD (with and without the C9ORF72 mutation) hippocampal pyramidal neurons were surrounded by coarse punctate staining, consistent with enlarged presynaptic terminals (I). Immunohistochemistry for TDP-43 (A–F), ubiquitin (G) and C9ORF72 (H and I). Scale bars: A, D–F, H and I = 25 µm; B and C = 30 µm; G = 12 µm.
Immunohistochemistry for ubiquitin and p62 labelled the compact TDP-43 immunoreactive neuronal cytoplasmic inclusions, dystrophic neurites and neuronal intranuclear inclusions but not the granular neuronal pre-inclusions or the small CA1 processes. A number of other types of inclusions were demonstrated with ubiquitin and p62 immunohistochemistry that were negative for TDP-43. These included small granular dot-like neuronal cytoplasmic inclusions and glial cytoplasmic inclusions and rare large swollen dystrophic neurites in the cerebral neocortex, focal cytoplasmic collections of granules in hippocampal pyramidal neurons, and neuronal cytoplasmic inclusions and small dystrophic neurites in the cerebellar granule cell layer (Fig. 4G). The most consistent and specific of these TDP-negative, ubiquitin/p62-immunoreactive pathologies was the cerebellar inclusions, which were present in all 18 study subjects examined but only rarely present in very small numbers in 25 mutation-negative FTLD-TDP and ALS controls.
In most cases, immunohistochemistry for tau and Aβ proteins did not show any specific pathological changes beyond those expected for patient age. However, three cases had sufficient numbers of classical neuritic senile plaques (CERAD ‘moderate’ or ‘frequent’) and neurofibrillary tangles (Braak stage V or VI) to fulfil diagnostic criteria for Alzheimer's disease (The National Institute on Ageing and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease, 1997). These cases were somewhat unusual in that all three showed a severe degree of cortical tau-immunoreactive neuritic pathology, disproportionate to the number of senile plaques, and a frequency of subcortical tau-immunoreactive neurites and glial cytoplasmic inclusions that was excessive for typical Alzheimer's disease (Supplementary Fig. 2). The frequency and anatomical distribution of the tau-immunoreactive neurites in these cases was distinct from the TDP-43 immunoreactive dystrophic neurites. No pathological changes were demonstrated with immunohistochemistry for α-synuclein or FUS.
Immunohistochemistry using a commercially available antibody against C9ORF72 showed weak cytoplasmic reactivity of most large neurons and diffuse finely punctate immunostaining of grey matter, consistent with synaptic localization. In cases with ALS, some lower motor neurons that appeared to be chromatolytic showed more intense cytoplasmic reactivity (Fig. 4H) and occasional swollen axons in the spinal cord ventral grey matter were strongly reactive. However, this staining pattern was also seen in cases with ALS in which the C9ORF72 mutation had been excluded. Hippocampal pyramidal neurons were surrounded by coarse punctuate staining, consistent with large presynaptic terminals (Fig. 4I). Although this pattern was more prominent than in neurologically normal controls, it was similar in other subtypes of FTLD and also seen in some cases with Alzheimer's disease. Most importantly, C9ORF72 immunohistochemistry did not demonstrate any cellular inclusions; specifically, it did not co-label the TDP-43 immunoreactive neuronal or glial inclusion and was not present in any of the TDP-negative, ubiquitin/p62-mmunoreactive neuronal inclusions.
Clinicopathological correlations
Cases with FTD had more severe degenerative changes and more TDP-43 immunoreactive pathology in the frontal and temporal neocortex than cases with ALS alone. However, the specific FTD subtype (behavioural variant FTD or PNFA) did not correlate with the lobar pattern (frontal versus temporal) of atrophy or degenerative change. Tissue blocks were not available from both right and left temporal lobes to assess possible correlation between laterality and language deficits. A diagnosis of ALS was associated with greater pyramidal tract degeneration and lower motor pathology. Moreover, the pattern of motor neuron disease showed the expected correlations; upper motor neuron dysfunction correlated with corticospinal tract degeneration while the degree of lower motor neuron dysfunction correlated with lower motor neuron degeneration and the frequency of TDP-43 immunoreactive inclusions. There was no correlation between the degree of extrapyramidal dysfunction and any measure of pathology in the striatum or substantia nigra. Memory loss was not associated with greater hippocampal pathology or the presence of concomitant Alzheimer's disease pathology. Interestingly, the presence of Alzheimer's disease and FTLD-TDP type A pathology both showed a positive correlation with age at death and were even more strongly associated with longer disease duration (P < 0.003).
Pathological variation within families
In four families (Families A, B, C and E), post-mortem examination had been performed on multiple affected members. Although there tended to be greater overlap in the neuropathology compared to the clinical phenotypes; nonetheless, there was significant heterogeneity among members of individual families, in the severity of degeneration and TDP-43 immunoreactive pathology in different neuroanatomical regions (Table 6). Moreover, two affected members of Family A showed different FTLD-TDP subtypes (only B versus B + A).
Discussion
Our findings confirm that the newly discovered C9ORF72 mutation is an important cause of familial FTD. In our series of cases, selected for the presence of the most common molecular subtype of FTLD (FTLD-TDP), the C9ORF72 mutation was found to be present in all autosomal dominant families in which multiple members were affected by a combination of FTD and ALS. In addition, all autosomal dominant families in our series with a pure FTD phenotype and FTLD-TDP pathology were explained by mutations in either C9ORF72 or GRN, with the two genetic abnormalities being of similar frequency in this group. We did not find mutations in any sporadic cases or those with only a weak family history; however, the number of such cases in our series was very small. The entry criteria of our study also prohibited us from determining the frequency of the C9ORF72 mutation in families or sporadic cases with clinically pure ALS. Nonetheless, our findings suggest that C9ORF72 is the most common genetic cause of familial FTLD-TDP and likely accounts for the majority of families with FTD/ALS.
As our series was centred in a dementia research clinic, there was probably a selection bias towards families with FTD. Despite this, over half of the subjects had ALS, providing us a broad view of the spectrum of clinical phenotypes associated with the C9ORF72 mutation. A striking finding was the degree of clinical heterogeneity, particularly at the time of initial presentation. The age of onset had a particularly broad range (40 years), while the disease duration also varied considerably (15 years). The initial diagnoses included behavioural variant FTD (37%), PNFA (13%) and ALS (30%). Significant clinical variation was also common within individual families. However, despite the heterogeneity in their initial presentations, a notable feature of our subjects was the degree to which the phenotypes converged as their disease progressed. Whereas all but one subject had a single diagnosis at the time of initial presentation, by the end of the disease course, 23% fulfilled diagnostic criteria for both FTD and ALS, 27% of those with FTD had features of both behavioural variant FTD and PNFA and 13% had a combination of ALS with both PNFA and behavioural variant FTD. In fact, the degree of overlap was even greater when milder symptoms were taken into account; 67% of those with ALS had at least some abnormalities of behaviour and/or language and 50% of those with FTD had some motor dysfunction. Although there was some variation in the pattern of motor neuron dysfunction, it was generally typical of classical ALS with mixed upper and lower motor neuron findings. Extrapyramidal dysfunction and memory problems were common but usually mild with late onset.
The spectrum of clinical features in our patients from families with the C9ORF72 mutation was similar to what has been reported previously in a number of chromosome 9p-linked families, all of which demonstrated a combination of FTD and ALS with marked clinical variability among family members (Momeni et al., 2006; Morita et al., 2006; Vance et al., 2006; Valdmanis et al., 2007; Luty et al., 2008; Le Ber et al., 2009; Gijselinck et al., 2010; Boxer et al., 2011; Pearson et al., 2011). However, we have shown that the C9ORF72 mutation may also be responsible for a significant proportion of families with a pure FTD phenotype. Distinguishing families with autosomal dominant FTD caused by the C9ORF72 mutation from those with either GRN or MAPT mutations may prove difficult on clinical grounds since these three groups show similar demographic features, frequencies of different FTD phenotypes and intrafamilial heterogeneity (Kelly et al., 2009).
In all cases, neuropathological assessment found TDP-43 immunoreactive inclusions in a broad range of neuroanatomical regions. Cases with FTD all had typical findings of FTLD-TDP while those with ALS all showed degeneration of both upper and lower motor neurons with TDP-43 immunoreactive neuronal cytoplasmic inclusions. Similar to the clinical phenotypes, there was significant overlap in the pathology between the major diagnostic groups; both cases with a final diagnosis of pure ALS had some TDP-43 immunoreactive pathology in the extramotor neocortex and hippocampus, while 10/11 (91%) cases with pure FTD had at least rare TDP-43 immunoreactive neuronal cytoplasmic inclusions in lower motor neurons (Table 6).
The cortical pattern of TDP-43 immunoreactive pathology in our subjects was always consistent with FTLD-TDP type B, which is the form that is most commonly found in patients with both FTD and ALS (Mackenzie et al., 2011). However, slightly more than a third of cases in our series also had pathological changes of FTLD-TDP type A. The presence of this dual pattern of FTLD-TDP was strongly associated with advanced age and longer disease duration. Interestingly, in Alzheimer's disease and many other common neurodegenerative conditions, TDP-43 immunoreactive pathology is found in 25–50% of cases as an age-related secondary pathological change (Arai et al., 2009). It usually has features of FTLD-TDP type A and is associated with the less common allele of the rs5848 polymorphism in the GRN gene (Dickson et al., 2010). This suggests that FTLD-TDP type B might be the primary pathology associated with the C9ORF72 mutation and that additional TDP-43 immunoreactive pathology develops in a genetically susceptible subset of patients with advancing age.
In three subjects, we also found sufficient numbers of neocortical senile plaques and neurofibrillary tangles to warrant a pathological diagnosis of Alzheimer's disease. Although this could simply represent the coincidental occurrence of a common age-related disorder in some of our older subjects (mean age, with Alzheimer's disease = 72.3 ± 3.5 versus without Alzheimer's disease = 58.5 ± 3.5), future studies are needed to explore whether or not the C9ORF72 mutation is associated with an increased risk for Alzheimer's disease. The fact that the Alzheimer's disease pathology was somewhat atypical, with more abundant tau-immunoreactive neuritic and glial inclusions, may indicate an influence of the C9ORF72 mutation on the development of Alzheimer's disease pathology. These cases also demonstrate that the finding of abundant tau-immunoreactive pathology in a case with clinical FTD does not necessarily restrict the diagnosis to FTLD-tau; there may still be coexistent TDP-43 immunoreactive pathology and an associated C9ORF72 mutation.
Finally, the most intriguing pathological finding in our subjects was the consistent presence of inclusions in certain neuronal populations, such as the granule cells of the cerebellar cortex, that were immunoreactive for ubiquitin and p62 but negative for TDP-43. Similar pathological changes have been described previously and seem to be specific for the spectrum of FTLD-TDP and ALS where they are more commonly found in familial cases (Pikkarainen et al., 2010; King et al., 2011). Our findings further suggest that this pathological change may be specifically related to the C9ORF72 mutation, which implies that this genetic abnormality results in the abnormal intracellular accumulation of some unknown protein(s) that is not TDP-43 or C9ORF72.
In summary, expansion of the GGGGCC hexanucleotide repeat in intron 1 of C9ORF72 is an important genetic cause of both FTD and ALS with TDP pathology and probably accounts for most families in which both conditions occur. Patients with this mutation show a high degree of heterogeneity in clinical presentation but the final phenotype usually includes a combination of frontotemporal lobar dysfunction and motor abnormalities. Discovery of the C9ORF72 mutation is the strongest evidence to date that FTD and ALS represent a clinicopathological spectrum of disease with overlapping molecular pathogenesis.
Funding
Canadian Institutes of Health Research (operating grants no. 179009, no. 74580 to I.R.A.M. and H.H.F.); Pacific Alzheimer's Research Foundation (centre grant C06-01 to I.R.A.M. and H.H.F.); ALS Association (to R.R.); National Institutes of Health (grants no. R01 NS065782, R01 AG026251 to R.R.) and Clinical Genetics Investigatorship award from the Canadian Institutes of Health Research (to G.-Y.R.H.).
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
We are grateful to the patients and family members who participated in this research and Drs B. L. Beattie, D. Foti and R. Stowe for providing clinical records on several patients. We thank Margaret Luk (research technologist, Vancouver General Hospital) for her excellent technical assistance.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- PNFA
progressive non-fluent aphasia


{kind=link}
{kind=link}
{kind=link}
{kind=link}