




Abstract
A single traumatic brain injury is associated with an increased risk of dementia and, in a proportion of patients surviving a year or more from injury, the development of hallmark Alzheimer’s disease-like pathologies. However, the pathological processes linking traumatic brain injury and neurodegenerative disease remain poorly understood. Growing evidence supports a role for neuroinflammation in the development of Alzheimer’s disease. In contrast, little is known about the neuroinflammatory response to brain injury and, in particular, its temporal dynamics and any potential role in neurodegeneration. Cases of traumatic brain injury with survivals ranging from 10 h to 47 years post injury (n = 52) and age-matched, uninjured control subjects (n = 44) were selected from the Glasgow Traumatic Brain Injury archive. From these, sections of the corpus callosum and adjacent parasaggital cortex were examined for microglial density and morphology, and for indices of white matter pathology and integrity. With survival of ≥3 months from injury, cases with traumatic brain injury frequently displayed extensive, densely packed, reactive microglia (CR3/43- and/or CD68-immunoreactive), a pathology not seen in control subjects or acutely injured cases. Of particular note, these reactive microglia were present in 28% of cases with survival of >1 year and up to 18 years post-trauma. In cases displaying this inflammatory pathology, evidence of ongoing white matter degradation could also be observed. Moreover, there was a 25% reduction in the corpus callosum thickness with survival >1 year post-injury. These data present striking evidence of persistent inflammation and ongoing white matter degeneration for many years after just a single traumatic brain injury in humans. Future studies to determine whether inflammation occurs in response to or, conversely, promotes white matter degeneration will be important. These findings may provide parallels for studying neurodegenerative disease, with traumatic brain injury patients serving as a model for longitudinal investigations, in particular with a view to identifying potential therapeutic interventions.
Introduction
While the acute effects of traumatic brain injury (TBI) can be devastating, there is growing evidence suggesting that TBI also initiates long-term neurodegenerative processes in a proportion of survivors (Johnson et al., 2012). Furthermore, TBI represents one of the strongest epigenetic risk factors for the development of Alzheimer’s disease as evidenced in epidemiological studies linking TBI with an increased incidence of Alzheimer’s disease (Mortimer et al., 1985, 1991; Sullivan et al., 1987; Gedye et al., 1989; Graves et al., 1990; Molgaard et al., 1990; O'Meara et al., 1997; Salib and Hillier, 1997; Schofield et al., 1997; Nemetz et al., 1999; Guo et al., 2000; Plassman et al., 2000; Fleminger et al., 2003).
TBI is known to induce a complex array of inflammatory responses in the acute post-injury phase, with initial blood–brain barrier compromise associated with infiltration of blood-borne polymorphonuclear leukocytes, T cells, macrophages and natural killer cells, both in human studies and in animal models (Holmin et al., 1995, 1998; Soares et al., 1995; Carlos et al., 1997; Holmin and Mathiesen, 1999; Stahel et al., 2000; Chen et al., 2004). In addition, resident, quiescent microglia have been shown to undergo both morphological and expressive changes following injury (Aihara et al., 1995; Gentleman et al., 2004; Wilson et al., 2004; Maxwell et al., 2006; Nagamoto-Combs et al., 2007) that contribute to the multitude of cytokines associated with post-traumatic inflammatory cascades (for review see Loane and Byrnes, 2010; Ziebell and Morganti-Kossmann, 2010). While early inflammation following TBI has long been viewed as an important contributor to pathology, it is increasingly recognized that aspects of acute inflammation may also promote important protective and regenerative effects (Lenzlinger et al., 2001; Browne et al., 2006; Ziebell and Morganti-Kossmann, 2010).
In contrast to the acute setting, there is very limited data on the temporal course of inflammation following TBI. Nonetheless, a limited autopsy series suggests that increased microglial activity may persist with long-term survival (Gentleman et al., 2004). More recently, PET imaging demonstrated increased [11C](R)PK11195 binding in survivors up to 17 years post-TBI, suggesting persistent microglial activation (Ramlackhansingh et al., 2011). Notably, inflammation is increasingly recognized as a chronic feature of neurodegenerative disease (Akiyama et al., 2000; Pratico and Trojanowski, 2000; Yoshiyama et al., 2007; Eikelenboom et al., 2010; Perry et al., 2010; Qian et al., 2010; Brettschneider et al., 2012a, b), with emerging experimental data indicating that it may be an early event (Yoshiyama et al., 2007). Thus, persistent neuroinflammation following injury may prove mechanistically important in the link between TBI and Alzheimer’s disease.
Due to the unique biomechanical nature of TBI, axons in white matter are highly susceptible to injury, making diffuse axonal injury one of the most common pathologies of TBI (Adams et al., 1982; Geddes et al., 1997, 2000). In particular, the corpus callosum is highly vulnerable during trauma (Adams et al., 1989; Graham et al., 1995a; Leclercq et al., 2001), with axonal pathology in the corpus callosum meeting the criteria for the lowest pathological grade of diffuse axonal injury (Adams et al., 1989). Moreover, recent studies suggest axonal pathology may persist for some time after injury in both humans (Chen et al., 2009) and animal models (Pierce et al., 1998; Chen et al., 2004), with damaged axons potentially serving as an important source of the Alzheimer’s disease-associated protein amyloid-β (Smith et al., 1999, 2003; Iwata et al., 2002; Stone et al., 2002; Chen et al., 2004, 2009; Johnson et al., 2010, 2012; Tran et al., 2011a, b). Neuroimaging studies also indicate that TBI may precipitate ongoing white matter degeneration (Gale et al., 1995). However, the underlying mechanisms driving this progressive loss of white matter remain unknown.
Here, we examine material from cases of TBI with survival times ranging from 10 h to 47 years for evidence of microglial reactivity when compared with age-matched, uninjured control subjects using immunocytochemistry for the microglial-associated markers CD-68 and CR3-43. In addition, evidence of ongoing white matter pathology including axonal degeneration and evidence of myelin breakdown is assessed.
Materials and methods
Cohort: demographic and clinical data
All tissues were obtained from the Glasgow TBI archive of the Department of Neuropathology, Southern General Hospital, Glasgow, UK. Tissue was acquired at routine diagnostic autopsy, and approval for its use was granted by the South Glasgow and Clyde Research Ethics Committee.
Three injury cohorts were selected for examination. Cohort 1 consisted of patients who died acutely after moderate/severe TBI, with survival <2 weeks (range 10 h–10 days; mean 2.4 days; n = 16). Cohort 2 consisted of patients who died in the sub-acute phase, defined as survival of 2 weeks–1 year (range 2 weeks–9 months; mean 86 days; n = 11). Cohort 3 comprised long-term survivors of TBI, with survival at least 1 year post-injury (range 1–47 years; mean 10.2 years; n = 25). This cohort was expanded and modified from recent publications (Johnson et al., 2011, 2012) to permit a temporal evaluation of pathology using tissue conducive to comprehensive image analyses. Detailed reports from the diagnostic autopsy and/or forensic reports were available for all and indicated a history of a single moderate/severe TBI, supported by the autopsy findings. None were in a persistent vegetative state before death. These TBI cohorts were compared with material from uninjured, age-matched control subjects (n = 44), with no documented history of TBI, Alzheimer’s disease or Down’s syndrome. Full clinical and demographic information for these cohorts, including cause of death, is provided in Table 1.
Clinical and demographic data for groups
| Group 1: acute TBI (n = 16) | Group 2: subacute TBI (n = 11) | Group 3: long-term TBI (n = 25) | Group 3: non-TBI controls (n = 44) | |
|---|---|---|---|---|
| Age, mean (range) (years) | 40.6 (9–75) | 36.5 (17–67) | 55.0 (19–89) | 47.0 (14–92) |
| <20 (%) | 4 (25) | 3 (27) | 1 (4) | 4 (9) |
| 20–39 (%) | 3 (19) | 3 (27) | 3 (12) | 16 (36) |
| 40–59 (%) | 7 (44) | 4 (36) | 14 (56) | 10 (23) |
| 60+ (%) | 2 (12) | 1 (9) | 7 (28) | 14 (32) |
| Males (%) | 11 (69) | 11 (100) | 21 (84) | 28 (64) |
| Mean survival time (range) | 2.4 days (10 h–10 days) | 86 days (2 weeks–9 months) | 10.2 years (1–47years) | Not applicable |
| Cause of TBI | ||||
| Assault (%) | 3 (19) | 3 (27) | 3 (12) | Not applicable—all control cases have no known history of TBI |
| Fall (%) | 9 (56) | 4 (36) | 11 (44) | |
| MVC (%) | 3 (19) | 4 (36) | 7 (28) | |
| Unknown (%) | 1 (6) | 0 (0) | 4 (16) | |
| Cause of death | ||||
| TBI (%) | 16 (100) | 2 (18) | 0 (0) | 0 (0) |
| Pulmonary (including bronchopneumonia/ARDS) (%) | 0 (0) | 7 (64) | 6 (24) | 6 (14) |
| Heart failure (%) | 0 (0) | 0 (0) | 5 (20) | 6 (14) |
| Sudden unexpected death due to epilepsy (%) | 0 (0) | 0 (0) | 4 (16) | 13 (30) |
| Acute cardiovascular death (%) | 0 (0) | 1 (9) | 3 (12) | 7 (16) |
| GI disease (including liver failure) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Drug overdose (%) | 0 (0) | 0 (0) | 0 (0) | 4 (9) |
| Malignancy (%) | 0 (0) | 0 (0) | 2 (8) | 2 (5) |
| Acute intracerebral haemorrhage (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Renal disease (including pyelonephritis) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Hypothermia (%) | 0 (0) | 0 (0) | 1 (4) | 1 (2) |
| Sepsis (%) | 0 (0) | 0 (0) | 0 (0) | 2 (5) |
| Volvulus (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| Myasthenia gravis (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| GSW (%) | 0 (0) | 1 (2) | ||
| Unknown (%) | 0 (0) | 1 (9) | 1 (4) | 0 (0) |
| Group 1: acute TBI (n = 16) | Group 2: subacute TBI (n = 11) | Group 3: long-term TBI (n = 25) | Group 3: non-TBI controls (n = 44) | |
|---|---|---|---|---|
| Age, mean (range) (years) | 40.6 (9–75) | 36.5 (17–67) | 55.0 (19–89) | 47.0 (14–92) |
| <20 (%) | 4 (25) | 3 (27) | 1 (4) | 4 (9) |
| 20–39 (%) | 3 (19) | 3 (27) | 3 (12) | 16 (36) |
| 40–59 (%) | 7 (44) | 4 (36) | 14 (56) | 10 (23) |
| 60+ (%) | 2 (12) | 1 (9) | 7 (28) | 14 (32) |
| Males (%) | 11 (69) | 11 (100) | 21 (84) | 28 (64) |
| Mean survival time (range) | 2.4 days (10 h–10 days) | 86 days (2 weeks–9 months) | 10.2 years (1–47years) | Not applicable |
| Cause of TBI | ||||
| Assault (%) | 3 (19) | 3 (27) | 3 (12) | Not applicable—all control cases have no known history of TBI |
| Fall (%) | 9 (56) | 4 (36) | 11 (44) | |
| MVC (%) | 3 (19) | 4 (36) | 7 (28) | |
| Unknown (%) | 1 (6) | 0 (0) | 4 (16) | |
| Cause of death | ||||
| TBI (%) | 16 (100) | 2 (18) | 0 (0) | 0 (0) |
| Pulmonary (including bronchopneumonia/ARDS) (%) | 0 (0) | 7 (64) | 6 (24) | 6 (14) |
| Heart failure (%) | 0 (0) | 0 (0) | 5 (20) | 6 (14) |
| Sudden unexpected death due to epilepsy (%) | 0 (0) | 0 (0) | 4 (16) | 13 (30) |
| Acute cardiovascular death (%) | 0 (0) | 1 (9) | 3 (12) | 7 (16) |
| GI disease (including liver failure) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Drug overdose (%) | 0 (0) | 0 (0) | 0 (0) | 4 (9) |
| Malignancy (%) | 0 (0) | 0 (0) | 2 (8) | 2 (5) |
| Acute intracerebral haemorrhage (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Renal disease (including pyelonephritis) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Hypothermia (%) | 0 (0) | 0 (0) | 1 (4) | 1 (2) |
| Sepsis (%) | 0 (0) | 0 (0) | 0 (0) | 2 (5) |
| Volvulus (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| Myasthenia gravis (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| GSW (%) | 0 (0) | 1 (2) | ||
| Unknown (%) | 0 (0) | 1 (9) | 1 (4) | 0 (0) |
ARDS = acute respiratory distress syndrome; GI = gastrointestinal; GSW = gunshot wound; MVC = motor vehicle collision.
Clinical and demographic data for groups
| Group 1: acute TBI (n = 16) | Group 2: subacute TBI (n = 11) | Group 3: long-term TBI (n = 25) | Group 3: non-TBI controls (n = 44) | |
|---|---|---|---|---|
| Age, mean (range) (years) | 40.6 (9–75) | 36.5 (17–67) | 55.0 (19–89) | 47.0 (14–92) |
| <20 (%) | 4 (25) | 3 (27) | 1 (4) | 4 (9) |
| 20–39 (%) | 3 (19) | 3 (27) | 3 (12) | 16 (36) |
| 40–59 (%) | 7 (44) | 4 (36) | 14 (56) | 10 (23) |
| 60+ (%) | 2 (12) | 1 (9) | 7 (28) | 14 (32) |
| Males (%) | 11 (69) | 11 (100) | 21 (84) | 28 (64) |
| Mean survival time (range) | 2.4 days (10 h–10 days) | 86 days (2 weeks–9 months) | 10.2 years (1–47years) | Not applicable |
| Cause of TBI | ||||
| Assault (%) | 3 (19) | 3 (27) | 3 (12) | Not applicable—all control cases have no known history of TBI |
| Fall (%) | 9 (56) | 4 (36) | 11 (44) | |
| MVC (%) | 3 (19) | 4 (36) | 7 (28) | |
| Unknown (%) | 1 (6) | 0 (0) | 4 (16) | |
| Cause of death | ||||
| TBI (%) | 16 (100) | 2 (18) | 0 (0) | 0 (0) |
| Pulmonary (including bronchopneumonia/ARDS) (%) | 0 (0) | 7 (64) | 6 (24) | 6 (14) |
| Heart failure (%) | 0 (0) | 0 (0) | 5 (20) | 6 (14) |
| Sudden unexpected death due to epilepsy (%) | 0 (0) | 0 (0) | 4 (16) | 13 (30) |
| Acute cardiovascular death (%) | 0 (0) | 1 (9) | 3 (12) | 7 (16) |
| GI disease (including liver failure) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Drug overdose (%) | 0 (0) | 0 (0) | 0 (0) | 4 (9) |
| Malignancy (%) | 0 (0) | 0 (0) | 2 (8) | 2 (5) |
| Acute intracerebral haemorrhage (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Renal disease (including pyelonephritis) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Hypothermia (%) | 0 (0) | 0 (0) | 1 (4) | 1 (2) |
| Sepsis (%) | 0 (0) | 0 (0) | 0 (0) | 2 (5) |
| Volvulus (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| Myasthenia gravis (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| GSW (%) | 0 (0) | 1 (2) | ||
| Unknown (%) | 0 (0) | 1 (9) | 1 (4) | 0 (0) |
| Group 1: acute TBI (n = 16) | Group 2: subacute TBI (n = 11) | Group 3: long-term TBI (n = 25) | Group 3: non-TBI controls (n = 44) | |
|---|---|---|---|---|
| Age, mean (range) (years) | 40.6 (9–75) | 36.5 (17–67) | 55.0 (19–89) | 47.0 (14–92) |
| <20 (%) | 4 (25) | 3 (27) | 1 (4) | 4 (9) |
| 20–39 (%) | 3 (19) | 3 (27) | 3 (12) | 16 (36) |
| 40–59 (%) | 7 (44) | 4 (36) | 14 (56) | 10 (23) |
| 60+ (%) | 2 (12) | 1 (9) | 7 (28) | 14 (32) |
| Males (%) | 11 (69) | 11 (100) | 21 (84) | 28 (64) |
| Mean survival time (range) | 2.4 days (10 h–10 days) | 86 days (2 weeks–9 months) | 10.2 years (1–47years) | Not applicable |
| Cause of TBI | ||||
| Assault (%) | 3 (19) | 3 (27) | 3 (12) | Not applicable—all control cases have no known history of TBI |
| Fall (%) | 9 (56) | 4 (36) | 11 (44) | |
| MVC (%) | 3 (19) | 4 (36) | 7 (28) | |
| Unknown (%) | 1 (6) | 0 (0) | 4 (16) | |
| Cause of death | ||||
| TBI (%) | 16 (100) | 2 (18) | 0 (0) | 0 (0) |
| Pulmonary (including bronchopneumonia/ARDS) (%) | 0 (0) | 7 (64) | 6 (24) | 6 (14) |
| Heart failure (%) | 0 (0) | 0 (0) | 5 (20) | 6 (14) |
| Sudden unexpected death due to epilepsy (%) | 0 (0) | 0 (0) | 4 (16) | 13 (30) |
| Acute cardiovascular death (%) | 0 (0) | 1 (9) | 3 (12) | 7 (16) |
| GI disease (including liver failure) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Drug overdose (%) | 0 (0) | 0 (0) | 0 (0) | 4 (9) |
| Malignancy (%) | 0 (0) | 0 (0) | 2 (8) | 2 (5) |
| Acute intracerebral haemorrhage (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Renal disease (including pyelonephritis) (%) | 0 (0) | 0 (0) | 1 (4) | 0 (0) |
| Hypothermia (%) | 0 (0) | 0 (0) | 1 (4) | 1 (2) |
| Sepsis (%) | 0 (0) | 0 (0) | 0 (0) | 2 (5) |
| Volvulus (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| Myasthenia gravis (%) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| GSW (%) | 0 (0) | 1 (2) | ||
| Unknown (%) | 0 (0) | 1 (9) | 1 (4) | 0 (0) |
ARDS = acute respiratory distress syndrome; GI = gastrointestinal; GSW = gunshot wound; MVC = motor vehicle collision.
Brain tissue preparation and immunohistochemistry
For all examinations, the intact brain was immersed in 10% formol saline at autopsy and fixed for at least 3 weeks before dissection. Sampling using a standardized protocol and paraffin embedding was as described previously (Graham et al., 1995b). Analyses were performed using 8 µm hemi-coronal sections of the parasagittal cortex at the level of the mid-thalamus to include the corpus callosum.
Single immmunohistochemcial labelling
Following deparaffinization and rehydration, sections were immersed in aqueous hydrogen peroxide (10 min) to quench endogenous peroxidase activity. Antigen retrieval was performed in a microwave pressure cooker, with subsequent blocking achieved using one drop of normal horse serum (Vector Labs) per 5 ml of Optimax buffer (BioGenex) for 30 min. Incubation with the primary antibodies was performed for 20 h at 4°C. Antibodies specific for the anti-human HLA-DP, DQ, DR antigen (CR3/43 clone) and the cluster of differentiation-68 (CD68) antigen (both Dako) were used at a concentration of 1:800 and 1:2000, respectively. Notably, both antibodies detect activated forms of microglia. Specifically, CR3/43 is a marker of major histocompatibility complex class II expressing activated microglia (Graeber et al., 1994), whilst CD68 detects a lysosomal glycoprotein expressed by microglia/macrophages (Greaves et al., 1998). In addition, serial sections were incubated with an antibody specific for the N-terminal amino acids 66–81 of the amyloid precursor protein (Millipore, 1:50 K). A biotinylated universal secondary antibody was then applied for 1 h (Vectastain Universal Elite kit, Vector Labs), followed by an avidin biotin complex as per the manufacturer’s instructions (Vectastain Universal Elite kit, Vector Labs). Finally, visualization was achieved using the 3, 3′-diaminobenzidine (DAB) peroxidase substrate kit (Vector Labs). Counterstaining with haematoxylin was performed, and sections were examined using light microscopy on a Leica DMRB microscope (Leica Microsystems).
Positive control tissue for immunohistochemistry included sections from cases with a history of Alzheimer’s disease with previously demonstrated activated microglia. Omission of the primary antibody was performed on the same material to control for non-specific binding.
Double immunohistochemical labelling
Following deparaffinization and rehydration of sections, antigen retrieval and blocking was performed as described earlier in the text. The anti-human HLA-DP, DQ, DR antigen (CR3/43 clone) (Dako) was applied overnight (4°C) at a concentration of 1:30. After rinsing, the Alexa Fluor 488 donkey anti-mouse IgG secondary antibody was applied for 2 h at room temperature. Anti-myelin basic protein (36–50) antibody produced in rat was subsequently applied at a concentration of 1:10 overnight (4°C), and the corresponding Alexa Fluor 594 donkey anti-rat IgG secondary antibody applied as above. Following rinsing, sections were coverslipped using fluorescence mounting medium (Dako) and imaged using a Zeiss LSM 710 confocal microscope.
Luxol fast blue/Cresyl violet staining for myelin
Following standard dewaxing and rehydration, tissue sections were immersed overnight in Luxol fast blue solution (Solvent Blue 38, Sigma) at 27°C. Sections were then immersed in 95% ethanol to remove excess stain before being rinsed in deionized water. Differentiation was initiated with immersion in 0.05% aqueous lithium carbonate for 10 s followed by differentiation in multiple immersions in fresh 70% ethanol until grey, and white matter could be distinguished and nuclei decolourized. After washing in deionized water, sections were immersed in cresyl violet solution for 5 min at 60°C and subsequently washed again in deionized water. Sections were then differentiated in 95% ethanol (with 100 µl per 300 ml of acetic acid) and then rinsed in 95% ethanol only. Finally, sections were dehydrated and coverslipped.
Analysis of immunohistochemical findings
All observations were conducted blind to the demographic and clinical information by two independent observers (V.J. and W.S.).
Activated microglia
CR3/43 stained sections from each case were scanned at ×20 using a Hamamatsu NanoZoomer 2.0-HT slide scanner, following which regions of interest for analysis were identified directly by annotation of these images using the Digital Image Hub (Slidepath) and associated software. Serial images of multiple overlapping fields for the annotated region of interest were then captured, exported and combined to provide a single composite image of the region of interest using Microsoft Image Composite Editor (Microsoft Research). This image was then exported to ImageJ (NIH), the background subtracted (rolling ball radius 50.0 pixels) and the Color Deconvolution plugin applied according to the haematoxylin/DAB setting (H DAB) to provide two separate images representing the counterstain and DAB immunostaining (Ruifrok and Johnston, 2001). This DAB-specific image was then thresholded, and the percentage area occupied by the identified positive staining calculated for the entire hemi-corpus callosum using standard algorithms in ImageJ.
In addition to these quantitative measures of area of immunoreactivity, the morphology of CR3/43 and CD-68 immunoreactive cells was also examined for classical microglial phenotypes and assessed semi-quantitatively as either displaying a predominance of cells with amoeboid morphology or with a predominantly ramified or mixed amoeboid/ramified morphology.
Extent and morphology of axonal pathology
The morphology distribution and pattern of axonal pathology was determined in amyloid precursor protein stained sections. The extent of axonal pathology within the entire hemi-corpus callosum was classified semi-quantitatively as follows: 0 = absent or minimal pathology (<10 immunoreactive profiles, defined as a single axonal bulb or varicosity); 1 = moderate pathology; and 2 = extensive pathology.
Measurements of corpus callosum thickness
Digital images captured as described earlier were viewed with the web-based application software, Slidepath Digital Image Hub (SlidePath) and measurements of the superior-to-inferior extent of the corpus callosum (thickness) obtained at three points along the mediolateral extent: at its medial extent at its juncture with the cingulate gyrus, at the midline of the corpus callosum and midway between these two points, with the corpus callosum thickness in each case taken as the mean of these three calculations.
Statistical analyses
Statistical analyses were performed using GraphPad Prism statistical software (GraphPad Software Inc). Pearson’s correlation testing was performed to determine any association between age and the percentage area of CR3/43 immunoreactivity identified using image analysis in all groups. The t-test was used to compare the percentage area of immunoreactivity between groups. The Fisher’s exact test was used to assess differences in the morphology patterns of immunoreactive cells between groups for both inflammatory cells and the semi-quantitative analyses of axonal pathology.
Results
Extent and morphology of CR3/43 and CD68 immunohistochemistry
Control subjects
As has been described previously (Mattiace et al., 1990; Sheng et al., 1998; Conde and Streit, 2006), there was marked age-dependent variability in the extent of activated microglia in control subjects. Specifically, increasing age was associated with increased percentage area of CR3/43 immunoreactivity throughout the corpus callosum, typically in a uniform distribution (R2 = 0.27; P = 0.0004). In addition, immunoreactive microglia in older control subjects frequently displayed morphological features in keeping with a degree of activation, namely thickened processes and larger cell bodies (Fig. 1A–C). However, overtly amoeboid cells were rarely observed, and in no cases were there a majority of immunoreactive cells with amoeboid morphology (Figs 1 and 2).
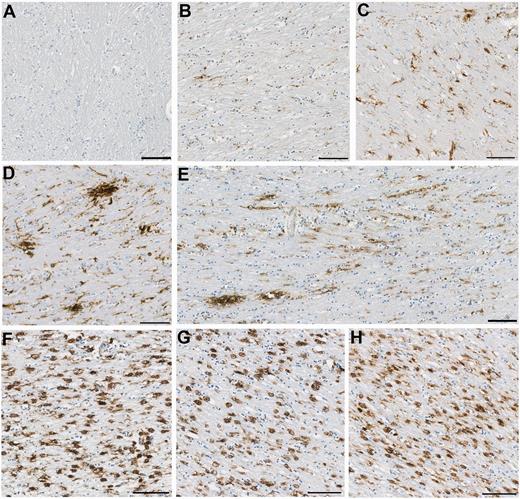
Representative images of observed CR3/43 immunoreactivity in the corpus callosum following TBI versus control subjects. (A–C) Representative images showing increasing CR3/43 reactivity with age as has been previously reported. (A) Virtually absent CR3/43 immunoreactivity in an 18-year-old female control subject who died as a result of leukaemia. (B) Minimal, highly ramified microglia observed in a 36-year-old female who died following a sudden cardiac event, and (C) numerous microglia with shortened, thickened processes and hypertrophy of the cell body, indicative of activation in a 92-year-old female who died as a result of bronchopneumonia. (D–E) Clusters of activated microglia with decreased ramifications in a (D) 23- and (E) 31-year-old male, who each died 4 weeks after TBI. Note the occasional cell displaying amoeboid morphology. In addition, cells can be seen arranging in parallel lines, likely along the length of an injured axon. (F–H) Extensive and densely packed amoeboid CR3/43 immunoreactive cells displaying minimal or no processes in (F) a 43-year-old male, 4 years post-TBI, (G) a 67-year-old male 8 months post-TBI and (H) a 64-year-old male 16 years post-TBI. All scale bars = 100 µm.
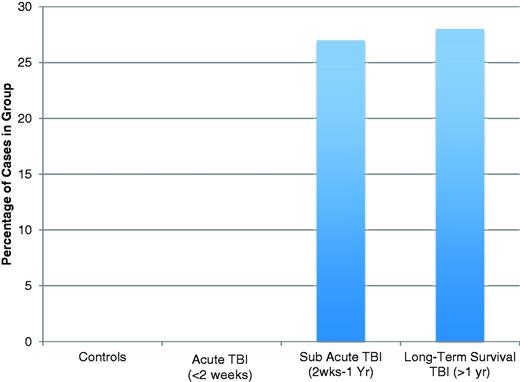
Percentage of cases displaying extensive amoeboid CR3/43 and CD68 immunoreactive cells in the corpus callosum following TBI by survival time versus control subjects.
Acute traumatic brain injury
In the acute phase following TBI, the mean percentage area of CR3/43 immunoreactivity did not differ from control subjects (3.37 ± 0.59% area stained in acute post-TBI versus 4.99 ± 0.5% in control subjects; P = 0.08). However, as in control subjects, an age-associated increase in immunoreactivity for microglia was noted (R2 = 0.32; P = 0.02). Further, at these early time points, cases with TBI displayed the same range of morphological features as control subjects, with only occasional microglia showing an amoeboid morphology, and no cases demonstrating a predominance of amoeboid cells (Fig. 2).
Sub-acute traumatic brain injury
During the sub-acute phase following trauma, there was evidence of increased microglia density and activity when compared with control subjects and acute cases. Specifically, the mean percentage area of immunoreactivity (for CR3/43) in the corpus callosum was 7.98 ± 2.01% in cases dying in the sub-acute phase following TBI versus 4.99 ± 0.5% in control subjects (P < 0.04) and 3.37 ± 0.59% in acute cases (P < 0.02). Moreover, the age-associated increase in microglia observed in control subjects and acute cases with TBI was not apparent subacutely (R2 = 0.05; P = 0.51). In this group, all but one case demonstrated CR3/43 and CD68 reactive cells with the typical morphological appearances of activated microglia, consistent with the previous literature (Engel et al., 2000), namely hypertrophied cell bodies and shortened, thickened ramifications (Fig. 1D and E). Specifically, in seven cases, intermediate cells with thickened processes and hypertrophied cell bodies falling short of amoeboid cells could be observed. However, in contrast to aged control subjects, these cells were not diffusely distributed, but appeared in dense clusters or linear arrangements (Fig. 1D and E). The remaining 3 of 11 (27%) cases demonstrated a predominance of microglia with overt amoeboid morphology densely packed throughout the corpus callosum (Figs 1G and 2). Notably, although numbers were small, the percentage area of CR3/43 immunoreactivity in this morphologically distinct subgroup was 13.80 ± 5.20, and substantially greater than control subjects (P = 0.0003) and acute cases (P = 0.0004).
Long-term survival
The percentage area of immunoreactivity in the long-term survival group was 4.63 ± 0.73% and was not statistically different from control subjects (P = 0.68), acute (P = 0.23) or sub-acute cases (P = 0.06). However, as in sub-acute cases, the age-associated increase in microglial activation observed in control subjects and cases with acute TBI was not observed in these long-term survival cases (R2 = <0.001; P = 0.93). Furthermore, although the area of immunostaining did not differ from control subjects, morphological examination revealed extensive regions predominantly populated by amoeboid microglia in over a quarter of cases (7 of 25; 28%) (Figs 1F–H, 2 and 3). These amoeboid microglia were observed over a wide survival range, from 2 to 8 years post-TBI. Typically amoeboid cells were immunoreactive for both CR3/43 and CD68 and were distributed throughout the corpus callosum, in contrast with the focal clusters of activated, yet ramified microglia observed in the sub-acute phase (Fig. 3). Notably, the percentage area of CR3/43 immunoreactivity in a subgroup of long-term survival cases that displayed a predominance of amoeboid cells was 8.65 ± 1.14, and substantially greater than control subjects (P = 0.0081), acute cases (P = 0.0002) and indeed long-term cases without morphological change (P = 0.0001).
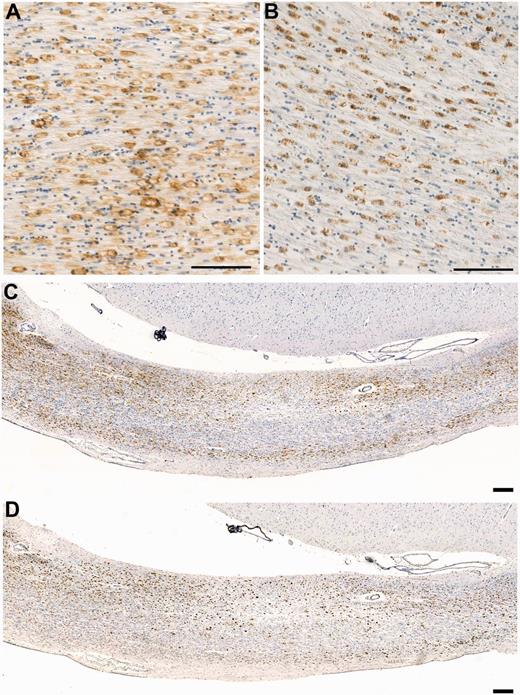
CR3/43 versus CD68 immunoreactivity in the corpus callosum in a case of long-term survival after TBI. (A) Dense CR3/43 reactive microglia with an amoeboid morphology in a 37-year-old male, 4 years post-injury. CR3/43 is an major histocompatibility complex Class II specific antibody and as such binds to the cell surface as can be observed. Scale bar = 100 µm. (B) The same region in this case displays a virtually identical cell population when subjected to immunohistochemistry specific for CD68. In contrast to CR3/43, CD68 immunoreactivity is predominantly intracellular, given that it is a lysosomal protein. Scale bar = 100 µm. (C–D) Low magnification photomicrographs displaying CR3/43 and CD68 immunoreactivity, respectively, in the same case as above. Cells tend to form bands along the inferior and superior border of the corpus callosum. Scale bar = 200 µm.
Interestingly, while cells were typically widely distributed throughout the corpus callosum, more densely populated bands of cells were commonly observed running the length of the inferior or superior aspect of the corpus callosum (Fig. 3) and were not observed in association with evidence of old or new lesions, such as haemorrhagic or ischaemic foci.
While numbers precluded formal statistical analyses, there appeared to be no association between the presence of extensive amoeboid cells and sex, cause of TBI or cause of death. The age of cases displaying predominantly amoeboid cells ranged from 28 to 89 years (mean 52.9). This is similar to the overall intermediate/long-term cohorts, and there was no overt age association.
Extent of axonal pathology
Axonal pathology was identified using immunohistochemistry to visualize amyloid precursor protein accumulating within axonal bulbs or swollen and tortuous varicosities along the length of damaged axons. The results indicate that, while axonal pathology diminished over time from the acute-injury phase (<2 weeks), evidence of ongoing axonal pathology was observed for up to 18 years post-trauma.
Control subjects
The majority of control subjects (35 of 44; 80%) displayed a complete absence or minimal axonal pathology (<10 axonal profiles) throughout the entire hemi-corpus callosum (score = 0) (Figs 4A and 5). The remaining nine controls (20%) displayed moderate–extensive axonal pathology (score 1–2) (Fig. 5). Notably, these controls frequently displayed axonal pathology in multiple wave-like bands widespread throughout the white matter (Fig. 4C), a pattern and distribution in keeping with hypoxic/ischaemic injury and likely to be indicative of prolonged episodes of hypoxia before death.
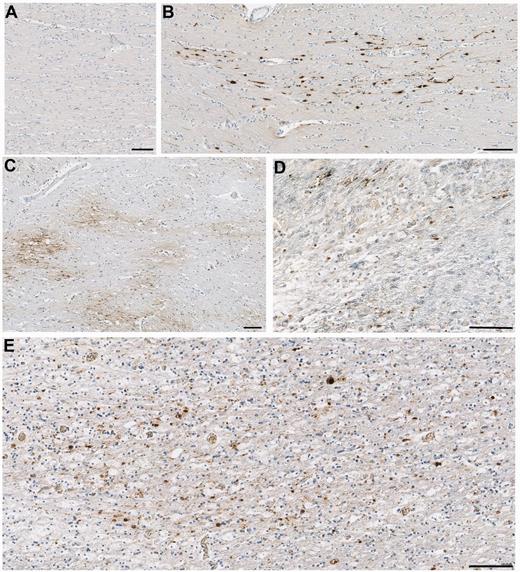
Representative images of axonal pathology in the corpus callosum as demonstrated by amyloid precursor protein immunoreactivity. (A) Normal white matter displaying no abnormal amyloid precursor protein immunoreactivity in a 67-year-old female who died following volvulus of the colon. (B) Axonal pathology with a distribution and morphology consistent with a traumatic origin in a 20-year-old male, 2 days post-injury. (C) Extensive axonal pathology in a pattern and distribution consistent with acute hypoxia-ischaemia in an 18-year-old male 10 h after TBI. (D) Small clusters of amyloid precursor protein immunonoreactivity consistent with the appearance of axonal bulbs in an 89-year-old female, 7 years post-TBI. On serial sections, the same region displayed extensive activated microglia as determined using CR3/43 and CD68 immunohistochemistry. (E) A similar pattern of multiple axonal bulbs in the corpus callosum of a 28-year-old male, 9 months post-TBI. Again, the same region displayed extensive activated microglia in serial sections. All scale bars = 100 µm.
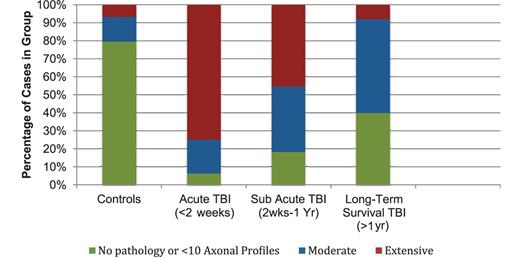
Extent of axonal pathology identified using amyloid precursor protein immunohistochemistry following TBI by survival time versus control subjects.
Cases with acute traumatic brain injury
As anticipated, axonal pathology was observed in all cases acutely following TBI and was greater than in control subjects (P < 0.001) (Figs 4B and 5). Pathology was extensive in 12 of 16 (75%) cases. Of the remaining four cases, three displayed moderate pathology (score = 1) and one case displayed minimal pathology (score = 0). Consistent with previous studies examining amyloid precursor protein immunoreactivity as a marker of axonal injury in TBI, pathology was identified at the earliest survival time point post-injury (10 h) (Gentleman et al., 1993; Sherriff et al., 1994).
Sub-acute traumatic brain injury
With survival from 2 weeks to 9 months following injury, the extent of axonal pathology, though less than in the acute survival group, remained considerably more prevalent and extensive than in control subjects (P > 0.001). Specifically, 5 of 11 (45%) cases displayed extensive pathology (score = 2), 4 of 11 (36%) had moderate pathology (score = 1) and just two cases had absent or minimal pathology (score = 0) (Fig. 5).
Long-term survival from traumatic brain injury
While axonal pathology was diminished with >1 year survival from TBI when compared with acute/sub-acute groups, the prevalence and extent of pathology remained considerably greater than in control subjects (P = 0.002). Specifically, while 2 of 25 (8%) cases had extensive pathology, 13 of 25 displayed moderate pathology (52%) and just 10 of 25 (40%) displayed absent or minimal pathology (Fig. 5).
Pattern, distribution and morphological appearance of axonal pathology with survival from traumatic brain injury
Within the first 2 months following TBI, axonal pathology was of a distribution and pattern indicating a mixed aetiology. This included isolated or small clusters of swollen axons in a single directional plane consistent with diffuse traumatic axonal injury (Fig. 4B), as well as more widespread waves of axonal pathology in keeping with the vascular complications of raised intracranial pressure or ischaemia (Fig. 4C). In contrast, in 13 cases with survival from 3 months to 18 years, a unique pattern, distribution and morphology of axonal pathology was observed, distinct from that of either acute traumatic axonal pathology or that associated with vascular complications. Specifically, isolated and somewhat granular axonal bulbs could be observed diffusely throughout the corpus callosum (Fig. 4D and E). By comparison, the long and tortuous varicosities classically described following acute TBI were rarely observed.
Notably, this more widespread axonal bulb pathology was highly correlated with abundant amoeboid neuroinflammatory cells observed on serial sections. Specifically, all 10 cases with overt neuroinflammation displayed axonal pathology, nine of which demonstrated this unique pattern and distribution of axonal damage. The remaining 10th case displayed extensive axonal pathology indicative of prolonged ischaemia that may have masked interpretation of more subtle axonal phenotypes.
White matter integrity
In all cases with TBI displaying extensive amoeboid microglia and axonal pathology, there was associated pallor of Luxol fast blue staining when compared with control subjects or cases with TBI with an absence of inflammation, regardless of survival time (Fig. 6). Luxol fast blue staining revealed not only a reduction in the overall density of staining for myelin but also structural and organizational disruption of the white matter. Where regions rich in cells identifiable morphologically as macrophages were noted, Luxol fast blue-positive fragments were frequently observed within the cytoplasmic compartment of cells, in keeping with active phagocytosis of myelin fragments. In further support of this, visualization of serial sections doublelabelled for CR3/43, and myelin basic protein using confocal microscopy confirmed myelin basic protein-immunoreactive fragments within cells immunoreactive for CR3/43 (Fig. 7).
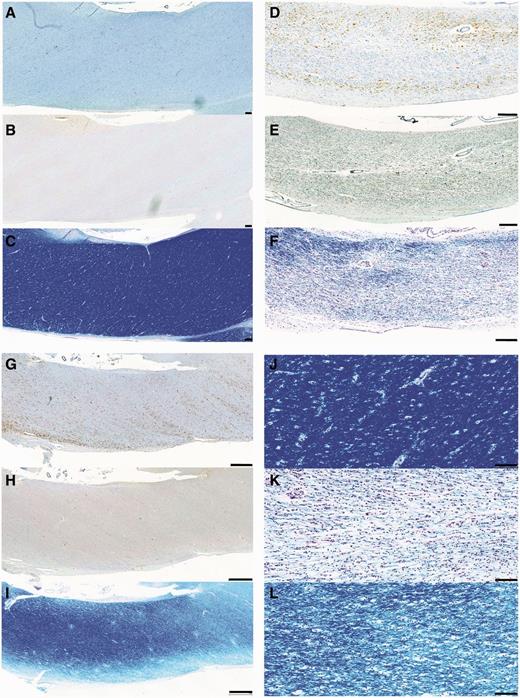
CR3/43, amyloid precursor protein and Luxol fast blue staining in the corpus callosum of representative cases with TBI versus control subjects. (A) CR3/43 (B) amyloid precursor protein and (C) Luxol fast blue staining in a 24-year-old male control subject who died following cardiomyopathy. Minimal CR3/43 immunoreactivity is accompanied by an absence of axonal pathology and white matter of a normal density and uniform distribution. Scale bars = 200 µm. (D) CR3/43 (E) amyloid precursor protein and (F) Luxol fast blue staining in a 37-year-old male who died 4 years post-TBI. Extensive amoeboid CR3/43 immunoreactivity is accompanied by multiple axonal bulbs and Luxol fast blue staining showing a decreased density of fibres and a non-uniform distribution of fibres within inflamed regions. Scale bars = 200 µm. (G) CR3/43 (H) amyloid precursor protein and (I) Luxol fast blue staining in a 43-year-old male who died 4 years post-TBI. Again, extensive amoeboid CR3/43 immunoreactivity is accompanied by minimal axonal pathology and a patchy loss of integrity of the white matter in association with regions of amoeboid microglia. Scale bars = 1 mm. (J) High magnification Luxol fast blue staining in the corpus callosum in the same case as A–C. (K) High magnification Luxol fast blue staining in the corpus callosum in the same case as D–F. (L) High magnification Luxol fast blue staining in the corpus callosum in the same case as G–I. Scale bars = 100 µm.
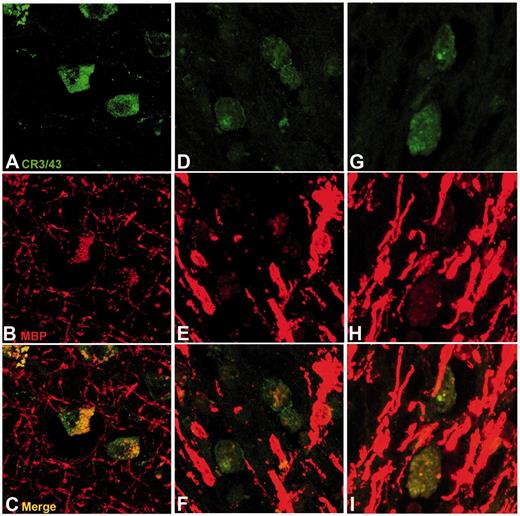
Representative images showing CR3/43 and myelin basic protein double labelling in the corpus callosum following survival from TBI. For all cases, CR3/43 is displayed in green and myelin basic protein (MBP) in red. (A–C) A 67-year-old male 8 months following TBI caused by a fall. (D–F) A 44-year-old female 2 years post-TBI caused by a fall. (G–I) A 37-year-old male 4 years post-TBI caused by a fall. Note the co-localization of myelin basic protein immunoreactivity within CR3/43 reactive cells, indicating phagocytosed myelin fragments.
Corpus callosum thickness
The thickness of the corpus callosum was reduced with survival of >1 year from TBI versus control subjects and acute survival cases (Figs 8 and 9). Specifically, the thickness of the corpus callosum was 2.97 ± 1.08 mm (SD) with long-term survival (>1 year) compared with 3.97 ± 1.17 mm in control subjects (P = 0.0008) and 3.74 ± 1.08 mm in acute cases (P = 0.03). Although there was a reduction in mean thickness between sub-acute cases (3.70 mm ± 1.05 mm) and long-term survivors, this did not reach statistical significance (t-test P = 0.48).
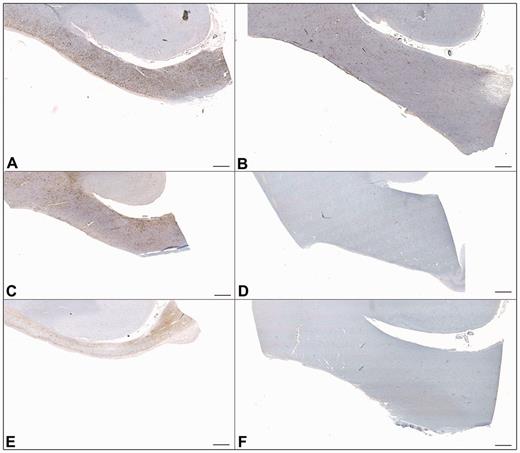
Representative low magnification images showing CR3/43 immunoreactivity in the hemi-corpus callosum following long-term survival from TBI versus representative age-matched control subjects. The thickness of the corpus callosum is markedly decreased following TBI. All images are presented at the same magnification and scale. All scale bars = 1 mm. (A) A 67-year-old male 8 months post-TBI compared with (B) a 60-year-old male control subject who died following heart failure. (C) A 44-year-old female who died 2 years post-TBI compared with (D) a 50-year-old male who died following bronchopneumonia. (E) A 37-year-old male who died 4 years post-TBI compared with (F) a 33-year-old female who died following an acute cardiac event.
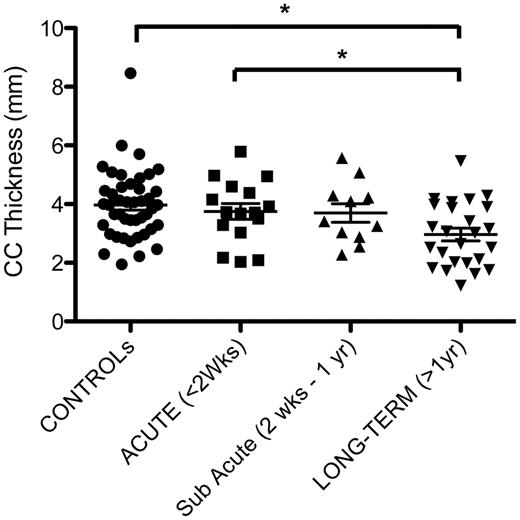
Thickness of the corpus callosum (CC) following survival from TBI versus uninjured control subjects. (* = p < 0.05).
Discussion
Here, we demonstrate that ongoing neuroinflammatory processes can persist in the corpus callosum many years after a single moderate-to-severe TBI in humans. Specifically, activated microglia and phagocytic macrophages were observed spanning extensive regions of the corpus callosum up to 18 years following injury. In addition, ongoing axonal degeneration and tissue atrophy were found in association with this inflammatory process. Potentially, chronic white matter inflammation contributes to the ongoing degeneration found here and with previous observations of progressive white matter pathology in human TBI and in animal models (Gale et al., 1995; Chen et al., 2004, 2009). Moreover, these observations may have important implications in the development of neurodegenerative conditions in TBI survivors, such as Alzheimer’s disease and chronic traumatic encephalopathy (Mortimer et al., 1985, 1991; Graves et al., 1990; Molgaard et al., 1990; O'Meara et al., 1997; Salib and Hillier, 1997; Schofield et al., 1997; Guo et al., 2000; Plassman et al., 2000; Fleminger et al., 2003).
As anticipated, within weeks after TBI, there was a predominance of microglia displaying somatic hypertrophy with thickening and shortening of processes, indicative of transformation to an activated phenotype. While cells with an overt macrophage phenotype could occasionally be observed in the sub-acute phase post injury, these were not abundant at this early time point. In contrast, with survival from TBI of ≥3 months, a proportion of cases began to display extensive regions consisting entirely of cells with an amoeboid morphology immunoreactive for CR3/43, a marker of major histocompatibility complex class II expressing activated microglia (Graeber et al., 1994), and CD68, a lysosomal protein expressed by microglia/macrophages (Greaves et al., 1998). As CD68 is associated with macrophages of a phagocytic phenotype (Greaves et al., 1998; Fleming et al., 2006), this would support the majority of cells comprising these regions being phagocytic macrophages. In 28% of cases with >1-year survival, this was the predominant cellular morphology of microglia in the white matter of the corpus callosum. Remarkably, this appearance could still be observed with survival of up to 18 years post-injury.
Notably, the extent of non-amoeboid CR3/43 and CD68 reactive microglia within control subjects demonstrated great variability. Although associated with age, as has been previously documented, the factors influencing microglial activation in the uninjured individual are acknowledged as numerous and complex (Perry et al., 2010). As such, interpreting the presence of large numbers of activated microglia post-trauma must be considered carefully in the unavoidably heterogeneous pathology of human TBI, thus highlighting the requirement for large series of cases and suitably matched uninjured control subjects in such studies. Notably, however, the presence of densely packed and predominant amoeboid cells within our cohort was unique to trauma cases surviving at least several months and was absent in both the control subjects and acutely injured cases. In addition, this pattern of pathology was observed across the age spectrum from 28 to 89 years old. While the numbers of cases here precludes any formal analysis of age association, the interplay of trauma, age and microglial activity will be an important topic for future research. In particular, the influence of an individuals’ baseline microglia activity may have important pathological and indeed functional consequences in both injury and repair.
Although the mechanisms driving this persistent inflammatory response post-TBI are unclear, its observation within the corpus callosum is of particular interest, as this represents a region highly susceptible to injury in TBI, with consequent axonal pathology (Adams et al., 1982; Geddes et al., 1997, 2000). It is of particular interest that dense regions of neuroinflammation were frequently observed extending laterally throughout the corpus callosum often along the inferior or superior border, perhaps reflecting regions under greatest biomechanical strain during the initiating traumatic insult. Studies to determine a potential relationship between mechanical strain and subsequent temporal patterns of inflammation will be important to examine either clinically or using clinically relevant models systems.
In association with the ongoing inflammation, amyloid precursor protein-immunoreactive, swollen axonal profiles were observed as solitary profiles or as clusters of somewhat granular axonal bulbs, indicative of disconnected axon terminals. This is in contrast to the long, varicose axonal profiles classically observed acutely post-trauma, presumed to represent multiple points of partial axonal transport interruption owing to traumatic axonal stretch or secondary ischaemic changes (Tang-Schomer et al., 2012). These findings suggest continued disconnection and degeneration of axons over time. In addition, long survival cases with TBI also displayed diminished or abnormal Luxol fast blue staining in keeping with loss of myelin, thus further supporting axonal loss through ongoing degeneration. Notably, double immunohistochemical labelling revealed material reactive for myelin basic protein within CR3/43 reactive cells indicating active phagocytosis. Interestingly, a marked reduction in thickness of the corpus callosum only became significant in survival of a year or more, post-TBI, supporting progressive loss from the initial insult, as opposed to an acute loss of tissue in the initial weeks following injury.
Together, these data are consistent with recent work demonstrating that axonal pathology may persist for some time following injury in both humans (Chen et al., 2009) and animal models (Pierce et al., 1998; Chen et al., 2004; Byrnes et al., 2012), in addition to radiological examinations indicating that TBI can induce selective white matter atrophy (Gale et al., 1995). Indeed, a recent MRI study by Tomaiuolo et al. (2012) indicates a chronic volume loss up to 8 years following TBI in the corpus callosum. Similar to our histopathological data, there was a difference between corpus callosum volume when imaged at ∼1 year versus 8 years post-TBI, indicating an ongoing atrophic process (Tomaiuolo et al., 2012). However, whether the chronic inflammation reported here is responsible for chronic axonal degeneration or, conversely, is induced in response to axonal degeneration, remains to be determined. Interestingly, a recent study in mice indicates that reduced microglia activation via the pharmacological activation of metabotropic glutamate receptor 5 (mGluR5) resulted in diminished white matter loss at 4 months post-injury with associated improvement in function (Byrnes et al., 2012). The ability to observe the temporal interplay between the potential beneficial and detrimental effects of the inflammatory response on the white matter will be an important future consideration.
Interestingly, a recent study by Ramlackhansingh et al. (2011) failed to show increased microglial activation in the corpus callosum of long-term TBI survivors as assessed by PET imaging using the ligand [11C](R)PK11195 (PK). Although various other brain regions displayed an increased PK-binding potential, notably remote from focal lesions, the corpus callosum displayed the lowest binding of all regions assessed. In addition, PK-binding failed to correlate with findings indicative of white matter injury as identified using diffusion tensor imaging. It is possible that differences in the patient population and the nature of injuries sustained may account for the discrepancies in identifying activated microglia between this study and the data presented here. Alternatively, it may be that the PK ligand, which targets a translocator protein expressed by the mitochondria of activated microglia, either failed to identify the same population of cells revealed by immunohistochemistry specific for CR3/43 and CD68 or failed to achieve sufficient resolution in vivo to identify the neuroinflammatory response noted here.
Although the role of inflammation in the pathophysiology of neurodegenerative disease is complex (Perry et al., 2010), the identification of active inflammation years after TBI is in a timeframe consistent with epidemiological associations linking TBI with the later onset of dementia (Mortimer et al., 1985, 1991; Graves et al., 1990; Molgaard et al., 1990; O'Meara et al., 1997; Salib and Hillier, 1997; Schofield et al., 1997; Guo et al., 2000; Plassman et al., 2000; Fleminger et al., 2003). Notably, axonal pathology has been implicated as a potential source of amyloid-β formation both acutely and long-term following TBI in humans and animal models (Smith et al., 1999, 2003; Iwata et al., 2002; Stone et al., 2002; Chen et al., 2004, 2009; Tran et al., 2011a, b). In addition, we have previously identified the hallmark Alzheimer’s pathologies of neurofibrillary tangles and amyloid-β plaques to a greater extent and, in the case of tangles, at a younger age in TBI survivors of a year or more versus control subjects (Johnson et al., 2012). Accordingly, it will be important to determine potential associations between inflammation and other neurodegeneration-associated proteins throughout the brain.
In summary, in this study, we demonstrate that a single moderate or severe TBI is associated with ongoing inflammation in the corpus callosum up to 18 years post injury. Cells with the immunohistochemical and morphological profile of macrophages were associated with increased axonal pathology and marked thinning of the corpus callosum. Together, these data indicate that TBI is capable of inducing a persistent neuroinflammatory pathology with associated white matter degradation. Further work to elucidate the underlying pathways driving this pathology and, in particular, whether this ongoing neuroinflammation is cause or effect, will be critical to our understanding of evolving pathologies post-TBI and may have relevance to our understanding of wider non-TBI associated neurodegenerative disease. Further, longitudinal studies on neuroinflammation in TBI may provide opportunities for targeted therapy development.
Funding
National Institutes of Health grants NS038104 (to D.H.S. and W.S.), AG038911 (to D.H.S.).
Acknowledgements
The authors would like to thank Ms Clare Orange, Pathology Unit, University of Glasgow who kindly assisted with digital slide scanning and the Think Pink Charity for providing financial support for the slide scanner.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}