




Abstract
Voltage-gated potassium channel complex antibodies, particularly those directed against leucine-rich glioma inactivated 1, are associated with a common form of limbic encephalitis that presents with cognitive impairment and seizures. Faciobrachial dystonic seizures have recently been reported as immunotherapy-responsive, brief, frequent events that often predate the cognitive impairment associated with this limbic encephalitis. However, these observations were made from a retrospective study without serial cognitive assessments. Here, we undertook the first prospective study of faciobrachial dystonic seizures with serial assessments of seizure frequencies, cognition and antibodies in 10 cases identified over 20 months. We hypothesized that (i) faciobrachial dystonic seizures would show a differential response to anti-epileptic drugs and immunotherapy; and that (ii) effective treatment of faciobrachial dystonic seizures would accelerate recovery and prevent the development of cognitive impairment. The 10 cases expand both the known age at onset (28 to 92 years, median 68) and clinical features, with events of longer duration, simultaneously bilateral events, prominent automatisms, sensory aura, and post-ictal fear and speech arrest. Ictal epileptiform electroencephalographic changes were present in three cases. All 10 cases were positive for voltage-gated potassium channel-complex antibodies (346–4515 pM): nine showed specificity for leucine-rich glioma inactivated 1. Seven cases had normal clinical magnetic resonance imaging, and the cerebrospinal fluid examination was unremarkable in all seven tested. Faciobrachial dystonic seizures were controlled more effectively with immunotherapy than anti-epileptic drugs (P = 0.006). Strikingly, in the nine cases who remained anti-epileptic drug refractory for a median of 30 days (range 11–200), the addition of corticosteroids was associated with cessation of faciobrachial dystonic seizures within 1 week in three and within 2 months in six cases. Voltage-gated potassium channel-complex antibodies persisted in the four cases with relapses of faciobrachial dystonic seizures during corticosteroid withdrawal. Time to recovery of baseline function was positively correlated with time to immunotherapy (r = 0.74; P = 0.03) but not time to anti-epileptic drug administration (r = 0.55; P = 0.10). Of 10 cases, the eight cases who received anti-epileptic drugs (n = 3) or no treatment (n = 5) all developed cognitive impairment. By contrast, the two who did not develop cognitive impairment received immunotherapy to treat their faciobrachial dystonic seizures (P = 0.02). In eight cases without clinical magnetic resonance imaging evidence of hippocampal signal change, cross-sectional volumetric magnetic resonance imaging post-recovery, after accounting for age and head size, revealed cases (n = 8) had smaller brain volumes than healthy controls (n = 13) (P < 0.001). In conclusion, faciobrachial dystonic seizures can be prospectively identified as a form of epilepsy with an expanding phenotype. Immunotherapy is associated with excellent control of the frequently anti-epileptic drug refractory seizures, hastens time to recovery, and may prevent the subsequent development of cognitive impairment observed in this study.
Introduction
The spectrum of immunotherapy-responsive clinical features associated with voltage-gated potassium channel (VGKC) complex antibodies has traditionally included isolated peripheral nerve hyperexcitability (Hart et al., 1997), Morvan’s syndrome (Liguori et al., 2001; Irani et al., 2012) and limbic encephalitis (Buckley et al., 2001; Schott et al., 2003; Thieben et al., 2004; Vincent et al., 2004). This VGKC-complex antibody associated limbic encephalitis is characterized by the acute to subacute onset of amnesia, disorientation and medial temporal lobe seizures. Although substantial recovery is seen after immunotherapy, patients often show residual amnestic deficits (Vincent et al., 2004; Chan et al., 2007).
Some patients with VGKC-complex antibody limbic encephalitis also have brief, very frequent seizures, typically involving posturing of the hemiface and ipsilateral arm (Irani et al., 2008). These events were commonly seen in the context of VGKC-complex limbic encephalitis and we proposed the term faciobrachial dystonic seizures to help clinicians recognize this distinctive and potentially treatable disorder (Irani et al., 2011). Faciobrachial dystonic seizures were consistently associated with antibodies against the LGI1 (leucine-rich, glioma inactivated 1 protein) component of the VGKC-complex and appeared to respond well to immunotherapy (Irani et al., 2008, 2011; Geschwind et al., 2008; Barajas et al., 2010; Andrade et al., 2011; Vincent et al., 2011; Quek et al., 2012). A recent multicentre study revealed that faciobrachial dystonic seizures often preceded the onset of limbic encephalitis, leading to the hypothesis that early immunotherapy for faciobrachial dystonic seizures might postpone or even prevent progression to the cognitive impairment that characterizes limbic encephalitis (Irani et al., 2011). However, this hypothesis was principally based on a series of VGKC-complex antibody-positive patients presenting with limbic encephalitis, in whom the presence of faciobrachial dystonic seizures was largely established retrospectively without serial cognitive assessments. Also, the phenotypic recognition was limited to the few preceding reported observations with potential for recall bias (Irani et al., 2008; Barajas et al., 2010).
Therefore, no study to date has (i) addressed the natural history of patients identified with faciobrachial dystonic seizures; (ii) described the phenotypic spectrum of the disorder; (iii) established definitively whether patients with this syndrome presented to neurologists without cognitive impairment; (iv) prospectively quantified the relative response to anti-epileptic drugs (AEDs) and immunotherapy; or (v) investigated whether treatments can influence the onset of cognitive impairment. Moreover, although brain MRI has shown that patients with radiological evidence of limbic encephalitis can progress to hippocampal atrophy (Urbach et al., 2006; Bien et al., 2007), it is not clear whether this would be the case after successful treatment of isolated faciobrachial dystonic seizures or observed in patients with VGKC-complex antibody limbic encephalitis without hippocampal signal change seen on clinical brain imaging. To address these questions, we undertook a prospective study over a 20-month period with longitudinal clinical, cognitive and serological assessments of 10 cases presenting with faciobrachial dystonic seizures to two UK neuroscience centres.
Materials and methods
Patient identification
Over 20 months (April 2010 to December 2011), as part of their routine clinical practice, the authors prospectively identified 10 cases in whom the likeliest presenting clinical diagnosis was faciobrachial dystonic seizures. Antibody results were not available when cases were diagnosed. Clinical data, serial Addenbrooke’s cognitive examination revised (ACE-R) scores (Mioshi et al., 2006), videos documenting the events, results of investigations including detailed serological assays (methods in Vincent et al., 2004; Irani et al., 2010a), and treatment responses, were ascertained under Oxfordshire Research Ethics Committee A approval (07/Q160X/28). Cases and their relatives also underwent detailed interviews and all consented to the publication of non-anonymized videos.
Magnetic resonance imaging and statistical analyses
To investigate the sequelae of their illness, eight individuals—seven from this cohort and another patient known to the authors with faciobrachial dystonic seizures but without cognitive impairment—consented (04/Q0406/147) to undergo a research scan including T1-weighted volumetric brain imaging using a 3 T Siemens Vario MRI scanner at the University of Oxford FMRIB centre during the convalescent phase of their illness. For all but one patient, data were acquired using a 32-channel receive coil using a multi-echo MPRAGE sequence (van der Kouwe et al., 2008), with resolution 1 × 1 × 1 mm, repetition time = 2530 ms, inversion time = 1200 ms, echo time = 1.69, 3.55, 5.41 and 7.27 ms, matrix size = 256 × 256 × 176, and GRAPPA factor = 2. For technical reasons, this case was scanned using a single-channel receive coil with a standard MPRAGE sequence with resolution 1 × 1 × 1 mm, repetition time = 2040 ms, inversion time = 900 ms, echo time = 4.7 ms, matrix size = 174 × 192 × 192 mm. Scans from an available sample of 13 healthy individuals (48 to 75 years of age) from multiple studies at the Dementia Research Centre were included for comparisons. These individuals had all undergone 3 T T1-weighted volumetric MRI on a Siemens TIM Trio 3 T using a standard MPRAGE sequence with isotropic 1.1 mm resolution (repetition time = 2200 ms, echo time = 2.9 ms, inversion time = 900 ms, field of view = 282 × 282 mm, matrix = 256 × 256, number of slices = 208). Volumetric image analysis was performed at the Dementia Research Centre at University College London. Images were corrected for intensity inhomogeneity using the N3 algorithm (Sled et al., 1998). Estimates of whole brain and hippocampal volume were produced through segmentations using BrainMAPS (Leung et al., 2011) and STEPS (Cardoso et al., 2011), respectively. Each segmented scan underwent a visual quality control process, with minimal manual editing where necessary. Total intracranial volume was generated by summing the grey matter, white matter, and CSF volumes as calculated using SPM8 (http://www.fil.ion.ucl.ac.uk/spm/software/spm8). This value was used to normalize the volume of the structures to head size. To allow for cross-sectional comparisons between individuals, brain volume and combined (left plus right) hippocampal volume were corrected for total intracranial volume. We calculated a hippocampal asymmetry index as the positive ratio of left minus right hippocampal volume to combined hippocampal volume × 100.
Results
Case-series summary: the spectrum of faciobrachial dystonic seizures
As summarized in Table 1 and in the online Supplementary Videos A–F, the features observed in the 10 prospectively identified cases with faciobrachial dystonic seizures included a broad adult age range (28–92 years), equal sex distribution, and stereotyped frequent events (8–200/day; median = 30) commonly triggered by emotions and movements, which were typically preceded by a sensory aura and accompanied by a disturbance of awareness. Other features strongly suggestive of epileptic seizures were the frequent ictal presence of automatisms (Supplementary Video D), and fear, agitation (Supplementary Video C) and speech arrest after the motor event. The motor component was always dystonic and involved the arm both proximally and distally (Supplementary Videos A–F). The ipsilateral face was spared in only two cases, whereas leg involvement was seen in almost half (40%) the cases. Events involving the leg alone were observed rarely in two cases in whom the majority of seizures also involved the face and arm. Events were typically brief but lasted between 10 and 30 s in four cases (Supplementary Video D). Interestingly, one patient had synchronous bilateral dystonia (Supplementary Video E) and two showed rapidly alternating events (Supplementary Video C, also seen in Kv1.1 knockout mice) (Smart et al., 1998). Three cases showed features consistent with alternative seizure semiologies: in one case versive head seizures preceded the faciobrachial dystonic seizures by many months and required two AEDs to achieve seizure control. Cases with faciobrachial dystonic seizures frequently had associated falls, often with serious injuries, and in one patient dystonic posturing of the arm and ipsilateral foot was observed during the fall.
Clinical characteristics of faciobrachial dystonic seizures in 10 cases
| Age and sex | 28–92 years (median 68 years); five female: five male |
| VGKC complex antibodies (n = 10) | Range = 346–4515 pM; mean = 1483 pM |
| LGI1 [n = 9; with CASPR2 (n = 1) and contactin-2 (n = 1)]. One negative for LGI1, CASPR2 and contactin-2 (VGKC-complex antibodies 377 pM). | |
| Faciobrachial dystonic seizures per day | 8–200 (mean = 68, median = 30) |
| Triggers (n = 8) | Heightened emotion (n = 5); kinesigenic (n = 2); loud noise (n = 2)a; concentration to conversations (n = 1) |
| Aura (n = 8) | Sensory symptoms (n = 7)b; auditory hallucination (n = 1) |
| Disturbance of consciousness | In eight cases |
| Motor events | |
| • Arm, face, leg involvement | In 10, eight and four cases, respectively |
| • Duration >10 s | In four cases |
| • Motor phenomenology | Dystonic (n = 10); clonic elements (n = 2) |
| • Only unilateral | In four cases |
| • Bilateral independent | In five cases – including rapidly alternating events in two cases |
| • Bilateral simultaneously | In one case |
| • Automatisms | In three cases |
| Falls (n = 7) | Injuries included fractured wrist (n = 1), ankle (n = 1) and subdural haematoma (n = 1) |
| After motor event (n = 5) | Fear and tearful (n = 1); agitation (n = 2)c; speech arrest (n = 4) |
| Other seizure semiologies (n = 3) | Head version with secondary generalization (n = 1); generalized tonic-clonic seizures (n = 2) |
| Age and sex | 28–92 years (median 68 years); five female: five male |
| VGKC complex antibodies (n = 10) | Range = 346–4515 pM; mean = 1483 pM |
| LGI1 [n = 9; with CASPR2 (n = 1) and contactin-2 (n = 1)]. One negative for LGI1, CASPR2 and contactin-2 (VGKC-complex antibodies 377 pM). | |
| Faciobrachial dystonic seizures per day | 8–200 (mean = 68, median = 30) |
| Triggers (n = 8) | Heightened emotion (n = 5); kinesigenic (n = 2); loud noise (n = 2)a; concentration to conversations (n = 1) |
| Aura (n = 8) | Sensory symptoms (n = 7)b; auditory hallucination (n = 1) |
| Disturbance of consciousness | In eight cases |
| Motor events | |
| • Arm, face, leg involvement | In 10, eight and four cases, respectively |
| • Duration >10 s | In four cases |
| • Motor phenomenology | Dystonic (n = 10); clonic elements (n = 2) |
| • Only unilateral | In four cases |
| • Bilateral independent | In five cases – including rapidly alternating events in two cases |
| • Bilateral simultaneously | In one case |
| • Automatisms | In three cases |
| Falls (n = 7) | Injuries included fractured wrist (n = 1), ankle (n = 1) and subdural haematoma (n = 1) |
| After motor event (n = 5) | Fear and tearful (n = 1); agitation (n = 2)c; speech arrest (n = 4) |
| Other seizure semiologies (n = 3) | Head version with secondary generalization (n = 1); generalized tonic-clonic seizures (n = 2) |
afaciobrachial dystonic seizures also kinesigenic in one case and triggered by concentration in another.
bascending tingling sensation across chest (n = 3), shoulder (n = 1) and face (n = 1, often with rising abdominal sensation), and cold (n = 1) or pain (n = 1) throughout body.
calso showed post-ictal speech arrest.
Clinical characteristics of faciobrachial dystonic seizures in 10 cases
| Age and sex | 28–92 years (median 68 years); five female: five male |
| VGKC complex antibodies (n = 10) | Range = 346–4515 pM; mean = 1483 pM |
| LGI1 [n = 9; with CASPR2 (n = 1) and contactin-2 (n = 1)]. One negative for LGI1, CASPR2 and contactin-2 (VGKC-complex antibodies 377 pM). | |
| Faciobrachial dystonic seizures per day | 8–200 (mean = 68, median = 30) |
| Triggers (n = 8) | Heightened emotion (n = 5); kinesigenic (n = 2); loud noise (n = 2)a; concentration to conversations (n = 1) |
| Aura (n = 8) | Sensory symptoms (n = 7)b; auditory hallucination (n = 1) |
| Disturbance of consciousness | In eight cases |
| Motor events | |
| • Arm, face, leg involvement | In 10, eight and four cases, respectively |
| • Duration >10 s | In four cases |
| • Motor phenomenology | Dystonic (n = 10); clonic elements (n = 2) |
| • Only unilateral | In four cases |
| • Bilateral independent | In five cases – including rapidly alternating events in two cases |
| • Bilateral simultaneously | In one case |
| • Automatisms | In three cases |
| Falls (n = 7) | Injuries included fractured wrist (n = 1), ankle (n = 1) and subdural haematoma (n = 1) |
| After motor event (n = 5) | Fear and tearful (n = 1); agitation (n = 2)c; speech arrest (n = 4) |
| Other seizure semiologies (n = 3) | Head version with secondary generalization (n = 1); generalized tonic-clonic seizures (n = 2) |
| Age and sex | 28–92 years (median 68 years); five female: five male |
| VGKC complex antibodies (n = 10) | Range = 346–4515 pM; mean = 1483 pM |
| LGI1 [n = 9; with CASPR2 (n = 1) and contactin-2 (n = 1)]. One negative for LGI1, CASPR2 and contactin-2 (VGKC-complex antibodies 377 pM). | |
| Faciobrachial dystonic seizures per day | 8–200 (mean = 68, median = 30) |
| Triggers (n = 8) | Heightened emotion (n = 5); kinesigenic (n = 2); loud noise (n = 2)a; concentration to conversations (n = 1) |
| Aura (n = 8) | Sensory symptoms (n = 7)b; auditory hallucination (n = 1) |
| Disturbance of consciousness | In eight cases |
| Motor events | |
| • Arm, face, leg involvement | In 10, eight and four cases, respectively |
| • Duration >10 s | In four cases |
| • Motor phenomenology | Dystonic (n = 10); clonic elements (n = 2) |
| • Only unilateral | In four cases |
| • Bilateral independent | In five cases – including rapidly alternating events in two cases |
| • Bilateral simultaneously | In one case |
| • Automatisms | In three cases |
| Falls (n = 7) | Injuries included fractured wrist (n = 1), ankle (n = 1) and subdural haematoma (n = 1) |
| After motor event (n = 5) | Fear and tearful (n = 1); agitation (n = 2)c; speech arrest (n = 4) |
| Other seizure semiologies (n = 3) | Head version with secondary generalization (n = 1); generalized tonic-clonic seizures (n = 2) |
afaciobrachial dystonic seizures also kinesigenic in one case and triggered by concentration in another.
bascending tingling sensation across chest (n = 3), shoulder (n = 1) and face (n = 1, often with rising abdominal sensation), and cold (n = 1) or pain (n = 1) throughout body.
calso showed post-ictal speech arrest.
There was a high rate of misdiagnosis, including among consultant neurologists: alternative diagnoses included a psychogenic movement disorder (n = 2) and an organic movement disorder most akin to paroxysmal kinesigenic dyskinesia (n = 1). Lacunar stroke (n = 1) and, in two cases with prominent falls, Stokes-Adams attacks were diagnosed by the admitting general physicians. Once memory loss was present, an early Alzheimer’s dementia was considered in three cases.
Investigation results
All cases had raised levels of serum VGKC-complex antibodies at initial testing (mean = 1483 pM, range 346–4515 pM, eight of ten > 400 pM). In nine of ten cases, the VGKC complex antibodies targeted LGI1 and from all available samples there was a good correlation between VGKC complex and LGI1 antibody concentrations (Fig. 1A; Spearman’s r = 0.74, P < 0.0001). One LGI1 antibody-positive case also had CASPR2 antibodies and another had contactin 2 antibodies. One patient with VGKC complex antibodies of 377 pM did not have LGI1, CASPR2 or contactin 2 specificities (Fig. 1A). VGKC complex antibodies sometimes fell to negative values (<100 pM) whereas LGI1-antibodies remained positive at low titres (Figs 1A and 5A).
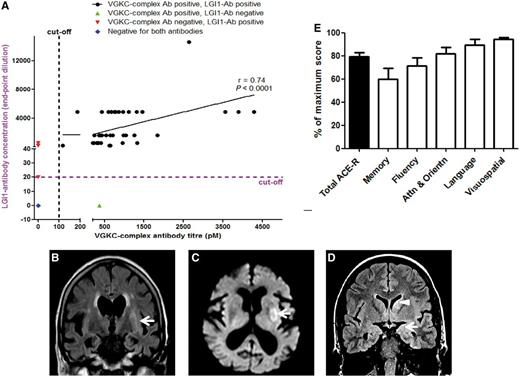
Serological and imaging findings in patients with faciobrachial dystonic seizures. (A) LGI1-antibody levels (measured by cell-based assay end-point dilution, Irani et al., 2010a) correlate with VGKC-complex antibody titres (measured by radioimmunoprecipitation assay, Vincent et al., 2004) (Spearman’s r = 0.74, P < 0.0001). Data shown for available samples from the nine cases with LGI1-antibodies. Some samples showed differences between the two assays (red and green triangles). (B) MRI brain showed left putaminal T2-weighted signal change (white arrow) and a corresponding area of restricted diffusion (C, white arrow) were observed in a 82-year-old female with faciobrachial dystonic seizures. (D) High signal in the left caudate (white arrowhead) and left medial temporal lobe (white arrow) were noted in a 28-year-old female with faciobrachial dystonic seizures and cognitive impairment. (E) Percentage of maximum ACE-R score (black bar) and category subscores (white bars) at time of maximal impairment (mean ± SEM). Ab = antibody; Attn & Orientn = attention and orientation.
By contrast to the strikingly consistent antibody results, the clinical brain MRI, body imaging and routine CSF analysis were usually unremarkable. When faciobrachial dystonic seizures alone were present, one of six cases showed abnormal routine clinical brain MRI, with high signal in the putamen (Fig. 1B and C). With cognitive impairment present, most conspicuously amnesia (Fig. 1E), only two of eight cases (both of whom developed generalized seizures) showed abnormalities that involved the medial temporal lobes (n = 2), and also the caudate in one (Fig. 1D). The basal ganglia signal changes were both contralateral to the faciobrachial dystonic seizures. In all seven cases where lumbar punctures were performed, cell counts, glucose, protein and oligoclonal band measurements were normal or negative. Serum sodium levels were reduced in all eight cases with cognitive impairment (Fig. 1E) but in only two of eight cases during periods with faciobrachial dystonic seizures alone (P = 0.007, Fisher’s exact test). No cases had clinical or neurophysiological (n = 4) evidence of peripheral nerve hyperexcitability. Ictal electrographic abnormalities were only observed in three cases, typically during longer clinical events. In two cases, focal slowing was observed during the faciobrachial dystonic seizures (Supplementary Fig. 1A and Supplementary Data). Immediately after a faciobrachial dystonic seizure, one other patient remained disorientated and showed limb and facial automatisms that were associated with rhythmic 4–5 Hz left-hemispheric discharges (Fig. 2 and Supplementary Video A). Interictal EEG showed diffuse slowing (n = 3), frequent subclinical right frontal and left temporal electrographic seizures (n = 1) and was normal in six. CT-PET/CT body performed in nine cases failed to disclose an occult tumour. One patient had a known familial cancer syndrome (multiple endocrine neoplasia type 1) with an active liver carcinoid syndrome and showed a marked improvement with immunotherapy (Fig. 5C).
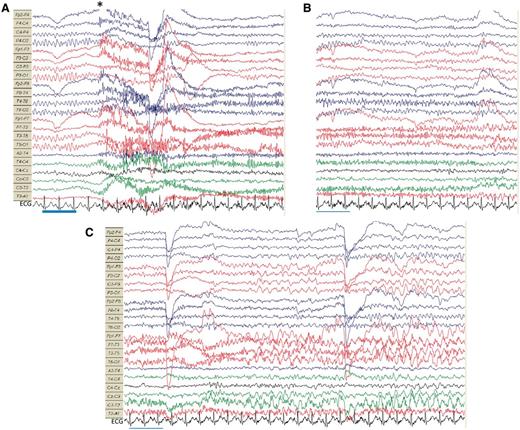
Electrographic changes associated with a faciobrachial dystonic seizure. (A) Symmetrical alpha rhythm is seen before the onset of myogenic artefact (*) associated with a right-sided faciobrachial dystonic seizure. Immediately afterwards, the patient was unresponsive to questioning. (B) Subsequently, there was relative attenuation of left hemisphere alpha rhythm, the patient remained disorientated with limb and orofacial automatisms. (C) Approximately 25 s after the faciobrachial dystonic seizure, rhythmic 4–5 Hz ictal discharge appears over the left hemisphere, maximal in the temporal region. Alpha rhythm normalizes after 45 s when the patient regains awareness. Blue time bar = 1 s. High frequency filter 35 Hz, low frequency filter 1 Hz. See Supplementary Video A.
Treatment of faciobrachial dystonic seizure spectrum events and associated side-effects
In all cases, AEDs were the initial treatment (median two AEDs, range 1–3), and were administered alone for between 11 and 200 days (median 29.5 days). During this period, in only one patient was the frequency of faciobrachial dystonic seizures reduced by >20% (Fig. 3A). AEDs were followed by corticosteroid administration in all cases with additional intravenous immunoglobulins (n = 4) and plasma exchange (n = 1). Within the first month of immunotherapy addition, >20% reduction in faciobrachial dystonic seizures was noted in nine cases (Fisher’s exact test P = 0.006). For each patient, the reduction in faciobrachial dystonic seizures after immunotherapy addition was recorded over the same time period for which they received AEDs only; there was a marked relative reduction in faciobrachial dystonic seizure frequency after immunotherapy addition to AEDs (Fig. 3A; Mann Whitney U test, P = 0.0001). Furthermore, cessation of the very frequent faciobrachial dystonic seizures was achieved in all cases after immunotherapy was added to AEDs and this often occurred rapidly after corticosteroid administration: within 7 days (n = 3), between 7 and 60 days (n = 6) and at 144 days (n = 1) (Figs 3B and 5A–D). The cessation of faciobrachial dystonic seizures was commonly observed before substantial reductions in serum VGKC-complex/LGI1 antibody titres (Fig. 5).
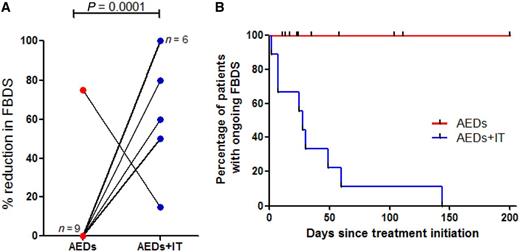
Clinical responses in patients with faciobrachial dystonic seizures. (A) The percentage reduction in faciobrachial dystonic seizures (FBDS) after anti-epileptic drugs (AEDs) alone (red) was significantly less when compared with the effect after addition of immunotherapy over the same time period (AEDs + IT, blue) (Mann Whitney U test P = 0.0001). In nine cases, AEDs alone had no impact on frequency of FBDS and in six cases after immunotherapy addition, there was complete cessation of faciobrachial dystonic seizures. (B) Only after addition of immunotherapy (IT) to AEDs was cessation of faciobrachial dystonic seizures observed.
Overall, AEDs caused a cutaneous reaction in a high proportion (50%) of cases (two phenytoin, two carbamazepine, one sodium valproate). In one case this also involved the eyes and mouth. Steroids were associated with psychiatric side-effects [paranoia (n = 2); hypomania (n = 3)], which consistently improved with tapering of steroids; one case suffered a spontaneous vertebral fracture (n = 1). Two cases noticed alteration of their typical seizure semiology immediately after steroids were commenced. One patient developed abnormal liver function tests (alanine transaminase of 1300 U/l) after intravenous immunoglobulins.
Immunotherapy may prevent progression from faciobrachial dystonic seizures to cognitive impairment
In this series, cases were identified solely on the basis of a semiology consistent with faciobrachial dystonic seizure-like events. When present, the onset of cognitive impairment with prominent amnesia (Fig. 1E), was often surprisingly insidious and accompanied by a significant increase in disability determined using the modified Rankin Score (Fig. 4A, Mann Whitney U, P = 0.008). The temporal relationship between the faciobrachial dystonic seizures and the development of cognitive impairment is summarized in Fig. 4B.
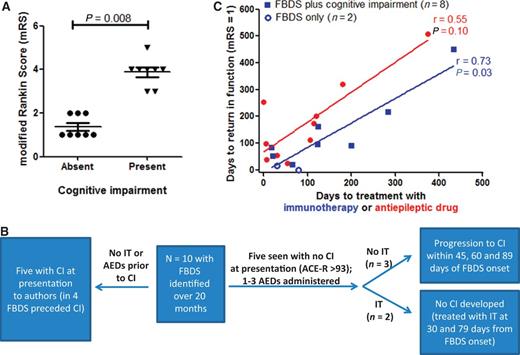
Prospective observation and treatment of faciobrachial dystonic seizures in 10 cases. (A) Modified Rankin score (mRS) was significantly lower in cases with faciobrachial dystonic seizures (FBDS) but without cognitive impairment (Mann Whitney U test, P = 0.008). (B) Absence of cognitive impairment (CI) was observed in cases administered immunotherapy (IT) while only faciobrachial dystonic seizures were present by comparison with cases offered no treatment or anti-epileptic drugs (AEDs) alone (P = 0.02, Fisher’s exact test). (C) The correlation between time to treatment with immunotherapy (blue) and time to return to baseline function (modified Rankin Score = 1; symptoms do not interfere with lifestyle) for cases with faciobrachial dystonic seizures with (n = 8, closed squares) and without (n = 2, open circles) cognitive impairment (P = 0.03, r = 0.74, Spearman’s correlation coefficient). Time to treatment with AEDs was not significantly correlated (P = 0.10, r = 0.55, red).
Five of the cases with faciobrachial dystonic seizures also had cognitive impairment when first seen; all had an ACE-R score <90. In these five, retrospective interviews revealed cognitive impairment preceded faciobrachial dystonic seizures in one (by 21 days) and appeared after the onset of faciobrachial dystonic seizures in four cases with a lag of between 12 h and 35 days. The other five cases were initially seen with faciobrachial dystonic seizures and no detectable cognitive impairment (ACE-R = 93–96). At this stage, all five cases were administered a variety of AEDs (median = 2; range 1–3) and three progressed to develop cognitive impairment at 45, 60 and 89 days, respectively. The two cases who did not develop detectable cognitive impairment (ACE-R = 94–99 throughout) were additionally given steroid therapy at days 30 and 79 into their illness. Therefore, all eight cases with faciobrachial dystonic seizures progressed to develop cognitive impairment without immunotherapy, despite treatment with AEDs in three. However, if immunotherapy was given when faciobrachial dystonic seizures alone were present, cognitive impairment was not detected (0/8 with no therapy or AEDs versus 2/2 with immunotherapy and faciobrachial dystonic seizures cessation; P = 0.02, Fisher’s exact test). VGKC-complex antibody levels did not differ between cases when cognitive impairment was present or absent (Supplementary Fig. 2).
All cases returned to their baseline function, although typically without complete normalization of formal neuropsychology testing scores (data not shown). Time to return to a modified Rankin Score of 1 (where symptoms do not interfere with lifestyle) was significantly correlated with time to immunotherapy administration (r = 0.74, P = 0.03), but not time to AED administration (r = 0.55, P = 0.10; Fig. 4C). The two cases that did not develop cognitive impairment showed the most rapid return to baseline function (Fig. 4C, blue open circles). Cases were followed for a median of 540 days (range 184–904).
Relapses of faciobrachial dystonic seizures
Relapses of faciobrachial dystonic seizures were seen in four of 10 cases (three shown in Fig. 5). During the relapse, no cases showed a change in their modified Rankin Score or ACE-R. Cases were either taking no prednisolone (Fig. 5A) or a low-dose tapering regime (Fig. 5B and C) and showed an absolute response, after 2–7 days, to an increase in corticosteroid dose without a change in AEDs. VGKC-complex/LGI1-antibodies persisted in all of these cases (Fig. 5A–C). Interestingly, the patient who stopped corticosteroids and AEDs once the antibodies were negative did not relapse. There were also two patients in who VGKC-complex/LGI1 antibodies returned without a relapse of faciobrachial dystonic seizures or cognitive impairment (Fig. 5D).
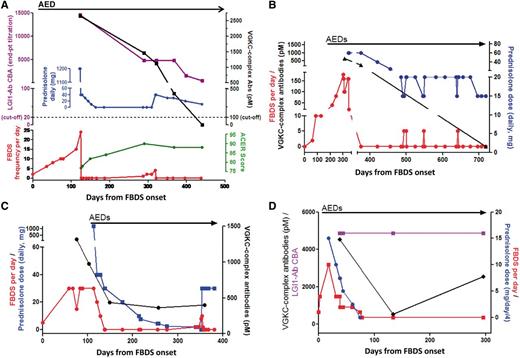
Clinical and serological features of corticosteroid-sensitive relapsing faciobrachial dystonic seizures (A–C) and persistent antibodies (D). Daily faciobrachial dystonic seizure frequency (FBDS, red), VGKC-complex antibodies (Abs; black), LGI1-antibodies measured by end-point titration of cell-based assay (LGI1-Ab CBA end-pt titration, purple), daily prednisolone dose (blue, mg, methylprednisolone equivalent calculated for first 3 days, divided by four in D), ACE-R and administration of AED(s). AED = anti-epileptic drug; ACE-R = Addenbrooke's cognitive examination-revised; CBA = cell-based assay.
Whole brain and hippocampal volumes
Eight patients consented to volumetric brain MRI which was performed after clinical improvement had plateaued (median 384 days). All eight had no temporal lobe signal change on clinical MRI during their illness; one showed left putaminal high-signal on FLAIR (Fig. 1B and C). There was no significant difference in age between cases (n = 8) and the 13 control subjects (70.6 ± 15.6 versus 64.4 ± 9.1, P = 0.25, Mann Whitney U test). Cases had significantly smaller combined hippocampal/total intracranial volume (3.03 ± 0.37 versus 3.70 ± 0.36, P = 0.002) and brain/total intracranial volume ratios (0.77 ± 0.05 versus 0.84 ± 0.04, P = 0.022) than control subjects. In regression analysis, there was a significant negative association between brain/total intracranial volume and increasing age for both cases and controls (Fig. 6A), but no association between combined hippocampal/total intracranial volume and age in either group (Fig. 6B). In joint regression analyses for the group as a whole, there were significant independent associations between brain/total intracranial volume and both age (P < 0.001) and case/control status (P = 0.001) (Fig. 6A). For combined hippocampal volume/total intracranial volume, there was evidence for a significant negative association with case/control status (P = 0.002) but not age (Fig. 6B). Furthermore, there were no differences noted when comparisons of brain/total intracranial volume and combined hippocampal volume/total intracranial volume were made between cases with and without cognitive impairment (Fig. 6C) or cases with greater or less than the median amount of total corticosteroid (7535 mg, Fig. 6D). Finally, all but one of the cases had an asymmetry index within the control range (0.3–5.4, median 2.7), the exception being one individual whose left hippocampus was 23% smaller than the right (Fig. 6E and outlier in Fig. 6F). This individual had exclusively right-sided faciobrachial dystonic seizures.
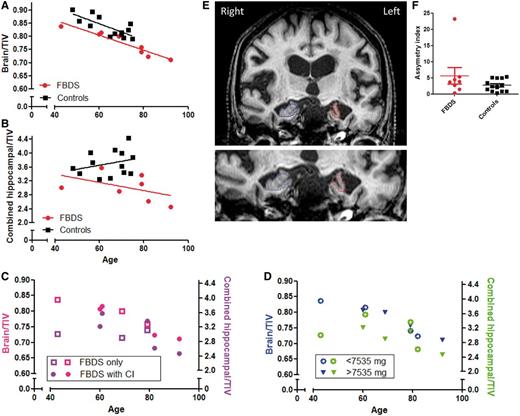
Volumetric hippocampal and whole brain imaging in patients with faciobrachial dystonic seizures. Correlation between age and brain (A) or combined hippocampal volume (×1000) (B) as a ratio of total intracranial volume (TIV) for controls (black) and cases with faciobrachial dystonic seizures (FBDS, red). (C) Total intracranial volume-normalized brain and combined hippocampal volumes in cases with (filled circles) and without (open squares) cognitive impairment (CI) and (D) in cases administered greater than (filled triangles) or less than (open circles) the median total corticosteroid dose (7535 mg). Two T1-weighted coronal MRIs through the left (red) and right (blue) hippocampi (E) of the case with marked hippocampal asymmetry (F).
Discussion
The presence of VGKC-complex antibodies has been well established in patients with peripheral nerve hyperexcitability, Morvan’s syndrome and limbic encephalitis (Hart et al., 1997; Liguori et al., 2001; Thieben et al., 2004; Vincent et al., 2004; Tan et al., 2008; Irani et al., 2012). This is the first prospective study of the—often exquisitely—immunotherapy-sensitive usually non-paraneoplastic syndrome of faciobrachial dystonic seizures, which establishes it as the fourth major syndrome consistently associated with VGKC-complex/LGI1 antibodies at levels similar to those seen in patients with limbic encephalitis (Vincent et al., 2004; Irani et al., 2010). During this study period only two VGKC-complex antibody limbic encephalitis patients without faciobrachial dystonic seizures were identified by the authors, suggesting that faciobrachial dystonic seizures are a frequent manifestation of VGKC-complex antibodies. If our experience is typical, a UK Neuroscience Centre might expect to see around four cases per year.
Previous accounts of VGKC-complex antibody positive patients (Irani et al., 2008, 2011; Barajas et al., 2010; Andrade et al., 2011; Aradillas and Schwarzmann 2011; Quek et al., 2012) described frequent, unilateral motor events with typical duration of <3 s which we termed faciobrachial dystonic seizures. Here, we advance the clinical characterization of faciobrachial dystonic seizures to include events in young adults and a patient in his nineties, attack durations of up to 30 s, synchronous, bilateral paroxysmal dystonic attacks, and a frequent association with a number of other well recognized epileptic features including sensory aura, manual automatisms, emotional and kinesigenic triggers, frequent disturbance of awareness, and speech arrest, fear or agitation after the dystonic event. Together with four other patients in whom ictal epileptiform EEG changes were seen (Andrade et al., 2011; Barajas et al., 2010), these observations strengthen the case for faciobrachial dystonic seizures being epileptic phenomena (contrast with Striano et al., 2011; Aradillas and Schwarzmann 2011). However, the ictal localization of faciobrachial dystonic seizures remains uncertain: scalp EEG changes are observed in the minority of events, the observed seizure semiologies are different from common frontal or temporal lobe semiologies, and basal ganglia imaging changes are surprisingly frequently observed (Irani et al., 2011). These observations may be most consistent with subcortical seizure involvement. Interestingly, auditory hallucinations were only noted in one case, making this a very different semiology from that seen in humans with LGI1 mutations in whom this is a common feature (Morante-Redolat et al., 2002). It is, however, notable that LGI1-deficient mice develop frequently asymmetrical dystonic or tonic postures (Chabrol et al., 2010).
Immunotherapy was associated with four impressive effects in this cohort. First, addition of immunotherapy to AEDs was associated with an often rapid and marked reduction in attacks that had hitherto been AED refractory. Notably, the complete cessation of faciobrachial dystonic seizures was only seen following corticosteroid addition. Second, relapses of faciobrachial dystonic seizures occurred after corticosteroid dose reductions, and repeat corticosteroid administration was associated with cessation of faciobrachial dystonic seizures within a few days without AED dose alteration. Third, in two of the five cases seen without cognitive impairment at presentation, corticosteroid administration was associated with an absence of subsequent cognitive impairment and disability which was observed in all eight cases given no treatment or AEDs alone. However, and given the small numbers involved, it is possible that some cases may never have been destined to develop cognitive impairment. Finally, time to administration of immunotherapy was significantly correlated with time to recovery of baseline function. This was not significant with time to AED administration although a similar trend was apparent. Taken together, these data provide strong evidence that immunotherapy, particularly when given early, may confer multiple benefits in the treatment of faciobrachial dystonic seizures, as has been shown in N-methyl-d-aspartate receptor antibody encephalitis (Dalmau et al., 2008; Irani et al., 2010b). In fact, it may be that prolonged courses of antibody-depleting immunotherapy are required in some patients as relapses of faciobrachial dystonic seizures were seen only in cases with persistent antibodies. Yet, the rapid clinical effect of corticosteroids often occurred before a significant fall in the antibodies. Furthermore, the immediate alteration of seizure semiology upon steroid administration in two cases, suggests that steroids are exerting, at least in part, an antibody-independent effect on faciobrachial dystonic seizures, perhaps through modulation of epileptic circuitry (Lalic et al., 2010), or through a reduction in parenchymal inflammation or blood–brain barrier permeability (Kashiwamura et al., 2011). Alternatively, the effect on CSF antibody concentrations may be more closely aligned with the clinical outcome, which was not measured here.
However, our data fall short of concluding that immunotherapy should be the initial treatment of choice in all patients with faciobrachial dystonic seizures. Corticosteroids were frequently associated with psychiatric side-effects in our cohort and are not without other risks in the elderly. Also, corticosteroids were always given after treatment with AEDs, raising the possibility of a synergistic effect of the two treatments.
Nevertheless, our data demonstrate that patients commenced on AEDs should be closely monitored for cutaneous side-effects and for disease progression (including the development of cognitive impairment) and the presence of either should lead to prompt immunotherapy. However, there have also been a few cases with faciobrachial dystonic seizures that have been reported to respond well to AEDs alone (Andrade et al., 2011; Irani et al., 2011; O’Connell et al., 2012).
The importance of immunotherapy may be extended by the presence of brain atrophy in cases with faciobrachial dystonic seizures with or without cognitive impairment. It is known that patients with VGKC-complex antibody limbic encephalitis and high signal in the medial temporal lobes typically progress to develop hippocampal atrophy as the high signal declines (Urbach et al., 2006; Bien et al., 2007, 2012). However, this has not been examined in VGKC-complex antibody positive cases without hippocampal signal change seen on clinical imaging. Our data suggest that patients with faciobrachial dystonic seizures in the convalescence stage do, at least on a group level, have smaller brain and hippocampal volumes than age-matched controls, despite the majority having had qualitatively normal clinical scans during their illnesses. This was seen in cases with and without cognitive impairment and did not appear to be influenced by the quantity of corticosteroid administered. These data are based on cross-sectional data from small numbers of subjects using a convenience sample of controls. Larger numbers and longitudinal imaging studies are required to quantify atrophy rates and determine the relationship between LGI1-antibodies, faciobrachial dystonic seizures frequency, immunotherapy and neurodegeneration. Nevertheless, these findings do provide evidence that the syndrome of faciobrachial dystonic seizures may produce deleterious effects both on hippocampal and whole brain structure. These diffuse effects are consistent with the known cortical and hippocampal distribution of LGI1 (Schulte et al., 2006), the presence of seizures in both regions in LGI1-deficient mice (Chabrol et al., 2010) and contrast with previous observations of hippocampal-specific atrophy (Urbach et al., 2006; Bien et al., 2007). It is notable that the only case with significant hippocampal asymmetry on structural imaging had exclusively contralateral faciobrachial dystonic seizures. Presuming the atrophy in this case was related to his illness, faciobrachial dystonic seizures may cause significant atrophy even in the absence of obvious signal change.
Given these therapy-related observations, it is important to note that not only were the faciobrachial dystonic seizures themselves commonly misrecognized by clinicians, including neurologists, but the subsequent encephalopathy was infrequently characterized by MRI, EEG and CSF changes that were previously reported in VGKC-complex antibody limbic encephalitis (Thieben et al., 2004; Vincent et al., 2004; Irani et al., 2010a; Lai et al., 2010). In addition to the frequent normality of investigations, the cognitive impairment was often insidious in onset. Therefore, this study may prompt an expansion in current diagnostic criteria of autoimmune encephalopathies to minimize underdiagnosis (Graus et al., 2004; Zuliani et al., 2012).
In some cases, where close antibody monitoring is considered useful, it may be worth measuring LGI1 antibodies rather than VGKC-complex antibodies as the latter were often low or negative while LGI1-antibodies were still detectable (Fig. 1A, red triangles). This may be particularly relevant for patients in remission who are considering withdrawal from immunotherapy and was also true for another patient at the onset of their illness. Conversely, at illness onset, one patient with faciobrachial dystonic seizures did not have LGI1, CASPR2 or contactin 2 antibodies despite VGKC-complex antibodies of 377 pM (Klein et al., 2012). Therefore, these two assays may offer complementary data as previously shown with CASPR2-antibodies (Becker et al., 2012).
Acknowledgements
We are grateful to Dr Katrina Morris and Mr Blake Hale for their help with data collection.
Funding
This work was supported by the National Institute for Health Research, Department of Health, UK (RDA/07/03/036 S.R.I.), ‘US-UK Fulbright Commission and the MS Society’ (S.R.I.), the Oxford NIHR Biomedical Research Centre (P.W., A.V.), Medical Research Council (P.P.), Imperial College Healthcare Trust Biomedical Research Centre (M.R.J.), and Epilepsy Research UK (S.R.I., B.L.).
Conflict of interest
A.V. and the Department of Clinical Neurology in Oxford receive royalties and payments for antibody assays and A.V. is the named inventor on patent application WO/2010/046716 entitled ‘Neurological Autoimmune Disorders’. The patent has been licensed to Euroimmun AG for the development of assays for LGI1 and other VGKC-complex antibodies. S.R.I., P.W. and B.L. are co-inventors and may also receive future royalties.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- ACE-R
Addenbrookes cognitive examination revised; AED = anti-epileptic drug; VGKC = voltage-gated potassium channel


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}