




Abstract
Mutations in the TPM2 gene, which encodes β-tropomyosin, are an established cause of several congenital skeletal myopathies and distal arthrogryposis. We have identified a TPM2 mutation, p.K7del, in five unrelated families with nemaline myopathy and a consistent distinctive clinical phenotype. Patients develop large joint contractures during childhood, followed by slowly progressive skeletal muscle weakness during adulthood. The TPM2 p.K7del mutation results in the loss of a highly conserved lysine residue near the N-terminus of β-tropomyosin, which is predicted to disrupt head-to-tail polymerization of tropomyosin. Recombinant K7del-β-tropomyosin incorporates poorly into sarcomeres in C2C12 myotubes and has a reduced affinity for actin. Two-dimensional gel electrophoresis of patient muscle and primary patient cultured myotubes showed that mutant protein is expressed but incorporates poorly into sarcomeres and likely accumulates in nemaline rods. In vitro studies using recombinant K7del-β-tropomyosin and force measurements from single dissected patient myofibres showed increased myofilament calcium sensitivity. Together these data indicate that p.K7del is a common recurrent TPM2 mutation associated with mild nemaline myopathy. The p.K7del mutation likely disrupts head-to-tail polymerization of tropomyosin, which impairs incorporation into sarcomeres and also affects the equilibrium of the troponin/tropomyosin-dependent calcium switch of muscle. Joint contractures may stem from chronic muscle hypercontraction due to increased myofibrillar calcium sensitivity while declining strength in adulthood likely arises from other mechanisms, such as myofibre decompensation and fatty infiltration. These results suggest that patients may benefit from therapies that reduce skeletal muscle calcium sensitivity, and we highlight late muscle decompensation as an important cause of morbidity.
Introduction
Skeletal muscle tropomyosin is one of the most abundant proteins in skeletal muscle, forming a key component of the thin filament that is intimately involved with the regulation of muscle contraction. Tropomyosin dimerizes into linear coiled-coil structures consisting of two α-helical chains. These dimers then bind in a head-to-tail manner to form continuous polymers that associate longitudinally along the length of actin thin filaments of muscle sarcomeres. Tropomyosin interacts with the troponin complex to regulate actin–myosin interactions and muscle contraction in response to cytoplasmic calcium (Ca2+) concentrations. There are three main tropomyosin isoforms in striated muscle. β-Tropomyosin, encoded by TPM2, is expressed in all skeletal muscle fibres and has low-level cardiac expression (Leger et al., 1976). TPM1 and TPM3 encode α-tropomyosinfast (Perry, 2001) and α-tropomyosinslow, respectively, which have a specific fibre-type expression (Pieples et al., 2000). These three tropomyosin genes, together with TPM4, also encode >30 cytoskeletal tropomyosin isoforms through the alternative splicing of various exons (Schevzov et al., 2005).
So far, mutations in TPM2 have been reported in association with four allelic disorders (Tajsharghi et al., 2012): nemaline myopathy (Donner et al., 2002), cap myopathy (Lehtokari et al., 2007), congenital fibre-type disproportion (Brandis et al., 2008) and distal arthrogryposis (Sung et al., 2003). Homozygous truncating mutations in TPM2 have been also associated with a distinctive form of nemaline myopathy with congenital large joint contractures and pterygia (Escobar syndrome) (Monnier et al., 2009). Although considered separate diagnostic entities, the presence of overlapping clinical and histological features in nemaline myopathy, cap myopathy and congenital fibre-type disproportion suggests that these disorders share similar disease mechanisms and belong to a continuous pathological spectrum. The defining pathological abnormality in nemaline myopathy is the presence of numerous nemaline bodies within myofibres that represent dense aggregates of Z-disc proteins such as actin and α-actinin. All mutations reported to date have been family-specific except for the p.E139del change, which has been reported in two families (Lehtokari et al., 2007; Clarke et al., 2009). Current evidence suggests that the phenotype associated with each TPM2 mutation is related to the specific effects of the mutation on protein function but our understanding of these relationships is far from complete (Tajsharghi et al., 2012). Mutations associated with chronic generalized muscle weakness have generally resulted in reduced affinity of the mutant β-tropomyosin for actin, reduced myofilament Ca2+ sensitivity or impaired cross-bridge cycling (Ochala et al., 2008; Marttila et al., 2012). Conversely, the congenital contractures associated with the p.R91G mutation likely arise because this mutation leads to an increase in myofilament Ca2+ sensitivity and possibly causes a hypercontractile phenotype in utero (Robinson et al., 2007). Other mutations give rise to both congenital contractures and ongoing weakness (Ochala et al., 2007). Currently, there are no therapies in clinical use that improve muscle performance and prognosis for congenital myopathies. Myofilament Ca2+ sensitizers and myosin activators are two drug classes that show promise for addressing abnormalities in sarcomere dynamics introduced by specific mutations. Indeed the Ca2+ sensitizer EMD 57033 improved the abnormal Ca2+ sensitivity associated with the p.E41K mutation (Ochala et al., 2008) and both CK-1909178 and EMD 57033 improved thin filament activation in TPM2-null and TPM3-R167H muscle biopsies (Ochala et al., 2012). Understanding disease mechanisms in different myopathies will be important to select the most appropriate medication and avoid complications in patients.
We report a novel recurrent heterozygous mutation (c.19_21delAAG; p.K7del) in the TPM2 gene associated with nemaline myopathy and/or core-rod myopathy in five unrelated families, making this the most common TPM2 myopathy found to date. We use a range of approaches to investigate the basis of the post-natal joint contractures. The TPM2 p.K7del mutation disrupts the ability of β-tropomyosin to incorporate into sarcomeres and progressive muscle contractures likely arise from a hypercontractile phenotype associated with an increased Ca2+ sensitivity of muscle contraction.
Materials and methods
Patient recruitment
Five unrelated families were identified by neuromuscular services in four different countries. Patient consent to reproduce clinical information was obtained from all participants, and laboratory studies were approved by the Human Research Ethics Committee of the Children’s Hospital at Westmead (CHW 2005/042).
Molecular modelling
We based the molecular modelling of human β-tropomyosin and the head-to-tail overlap region of β-tropomyosin dimers on the X-ray structure of pig α-tropomyosinfast (Protein Data Bank accession code 1C1G) (Whitby et al., 2000) in combination with the previously determined end-on-end dimer associations observed in both rat and chicken α-tropomyosin peptides (Protein Data Bank accession codes 2G9J and 3MUD, respectively) (Greenfield et al., 2006; Frye et al., 2010; Li et al., 2011). The University of California, San Francisco (UCSF)-chimera molecular graphics program (Pettersen et al., 2004) was used for molecular modelling.
Immunohistochemical and histological analysis
Muscle biopsies were snap frozen in isopentane cooled in liquid nitrogen. Biopsies were analysed from cryosections processed using standard histochemical stains. The morphometry analysis was performed on at least 200 fibres using ImagePro Plus 4 software (Media Cybernetics). Methods for analysis of DNA fragmentation, a marker of apoptosis, using the terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay are included in the Supplementary material.
Western blotting and 2D polyacrylamide gel electrophoresis
Western blotting was based on methods previously described (Cooper et al., 2003). For all proteins except tropomyosin, we used pre-cast gradient 4–12% NuPAGE™ Novex Bis–Tris gels (Invitrogen, Life Technologies). To resolve the different sarcomeric tropomyosin isoforms, we used 4/9% discontinuous gels containing 3.4 M urea in the resolving phase. The same gels were used for 2D PAGE for tropomyosin. We used the following primary antibodies: TPM311, which recognizes sarcomeric tropomyosin isoforms (mouse, 1:20 000; Sigma-Aldrich) and γ9d, which recognizes cytoskeletal tropomyosin isoforms that contain 9d exons (kindly provided by Prof. Peter Gunning and Dr Gallina Schevzov). Methods for western blotting of porin and mitochondrial oxidative enzyme complexes are included in the Supplementary material. Isoelectric focusing for tropomyosin was based on methods previously described (Ilkovski et al., 2004). In the first dimension, 18-cm immobiline dry strips (pH 4.5–5.5, Amersham Biosciences, GE Life Sciences) were focused using a Multiphor™ IEF unit (Amersham Biosciences) for a total of 61 kV/h (gradient mode starting at 0.5 kV, increasing gradually to 3.5 kV). Tris-glycine PAGE was performed on purified wild-type β-tropomyosin (WT-βTm) and K7del-β-tropomyosin (K7del-βTm) recombinant proteins (at ∼2 mg/ml) with and without SDS present in the loading buffer.
Cell culture models
We generated constructs for the WT-βTm-enhanced green fluorescent protein (EGFP) and the p.K7del-βTm-EGFP using the human muscle-specific isoform of TPM2 amplified from control muscle complementary DNA and an EGFP plasmid containing a kanamycin cassette. Further details are included in Supplementary material. C2C12 cell culture, transfection and differentiation were performed as previously described (Ilkovski et al., 2004). Primary myoblast and fibroblasts cell lines were established using the explant culture method (Decary et al., 1997). Once established, primary human myoblasts were routinely grown in Dulbecco’s modified Eagle medium:Ham’s F12 (1:1 ratio), 10% v/v amniomax, 20% v/v foetal bovine serum, 40 μg/ml gentamicin. All cell culture media and products were from Invitrogen unless indicated.
Immunohistochemistry of cultured cells
Cells grown on collagen/MatrigelTM-coated plastic Thermanox coverslips (Nunc) were fixed and permeabilized in PBS solution containing 3% v/v paraformaldehyde and 0.1% v/v Triton™ X-100 for 20 min at room temperature. Immunocytochemistry was performed as previously described (Ilkovski et al., 2004). Imaging was undertaken using a Leica TCS SP2 scanning confocal microscope. Primary antibodies used recognize sarcomeric actin (5C5 mouse Ab, 1:2000, Sigma-Aldrich) and α-actinin 2 (4B3 rabbit Ab, 1:500 000, kindly provided by Prof. Alan Beggs, Harvard Medical School, Boston, MA, USA). We used Cy3-conjugated goat anti-mouse IgG (1:250, Jackson ImmunoResearch Laboratories Inc) and Alexa Fluor® 488-conjugated goat anti-rabbit IgG (1:250, Molecular Probes) secondary antibodies. TO-PRO®-3 iodide (1:200, Molecular Probes, Life Technologies) was incubated with the secondary antibody to stain nuclei.
Extraction of sarcomeric and non-sarcomeric tropomyosin protein pools
A centrifugation method was used to separate tropomyosin bound in high molecular weight structures (insoluble pool representing protein within sarcomeres and nemaline bodies) and unbound protein (soluble) as previously described (Domazetovska et al., 2007). For preparation of insoluble and soluble protein pools for 2D-PAGE analysis, the primary cultured myotubes were washed twice with cold PBS and collected by washing the wells with extraction buffer [50 mM MES pH 6.8, 1 mM EGTA pH 8.0, 50 mM KCl, 1 mM MgCl2, 0.5% v/v Triton™ X-100, protease inhibitor cocktail (1:500, Sigma)]. This was followed by ultracentrifugation at 100 000 g for 1 h. The supernatant, representing the soluble protein fraction, was transferred to a fresh tube, and the pellet, representing the insoluble fraction, was resuspended in 150 μl rehydrating solution [2% v/v Triton™ X-100, 8 M urea, 2% v/v IPG buffer pH 4–7 (Amersham Biosciences), 36 mM DTT, protease inhibitor cocktail (1:500)]. The insoluble fraction was sonicated (eight bursts) and incubated with Lambda protein phosphatase (Lambda PP-P0753S, New England Biolabs) to dephosphorylate protein samples. To separate insoluble and soluble pools in patient muscle, a low-speed centrifugation protocol was used (13 000g) after brief solubilization of 8-μm thick cryosections in a buffer containing 0.5% v/v Triton™ X-100 as described previously (Ilkovski et al., 2004).
RNA isolation and amplification of the sarcomeric and cytoskeletal TPM2 gene isoforms
RNA isolation from patients’ primary cultured myotubes and fibroblasts was performed using the TRI Reagent® method (Molecular Research Centre) as per the manufacturer’s instructions, followed by ethanol precipitation. Complementary DNA was then synthesized using the oligo-dT15 method (Invitrogen). The β-tropomyosin complementary DNA sequence was amplified using isoform-specific primers. The TM1 cytoskeletal transcript (also transcribed from TPM2) was amplified from patient fibroblasts using similar methods. Primer sequences are available on request.
Synthesis of β-tropomyosin in Sf9 insect cells
We used site-directed mutagenesis to generate both K7del-βTm and WT-βTm viruses to infect Sf9 insect cells using the baculovirus expression method as described previously (Akkari et al., 2002; Marttila et al., 2012). Tropomyosin synthesized using this system undergoes N-terminal acetylation, which is important for tropomyosin dimer end-on-end association.
In vitro motility assay
We compared the effects of recombinant WT-βTm and K7del-βTm on the sliding speed of reconstituted thin filaments using the in vitro motility assay, which measures the Ca2+-regulation of movement of single thin filaments over immobilized myosin (Fraser et al., 1995; Bing et al., 1997). See Supplementary material for details.
Studies of patient single myofibres
To investigate the effects of the p.K7del mutation on sarcomeric dynamics, we directly measured a range of contractile properties in myofibres derived from patient muscle biopsies. In brief, we dissected individual myofibres from frozen muscle biopsies from two unrelated patients heterozygous for the TPM2 p.K7del mutation and controls using methods described previously (Ottenheijm et al., 2009, 2010, 2011). Fibres were mounted between a length motor and a force transducer element using aluminium T-clips and were activated using solutions with different Ca2+ concentrations. The maximum specific force and the sensitivity of myofibre force to Ca2+ were determined.
Actin–tropomyosin binding assay
We investigated the effects of the p.K7del mutation on β-tropomyosin–actin binding using an actin–tropomyosin co-sedimentation assay based on previously described methods (Mirza et al., 2007) and an actin-binding kit (Cytoskeleton; Cat. # BK001). Filamentous actin (18 µM) was incubated with varying amounts of K7del-βTm or WT-βTm (for 30 min at room temperature and co-sedimented at 150 000g). Pellets and supernatants were analysed by discontinuous 5%/12% SDS-PAGE, and data were fitted to the Hill equation to determine the dissociation constant (Kd) and Hill’s coefficient (h) using Prism 5.01 (GraphPad Software). See Supplementary material for further details.
Biophysical analyses of K7del-βTm structure
To investigate the effects of the p.K7del mutation on the ability to form homodimers, the structure of homodimers and the ability to polymerize, we performed circular dichroism (CD), small-angle X-ray scattering (SAXS) and native PAGE analysis of recombinant WT-βTm and K7del-βTm. Proteins first underwent purification by hydroxyapatite chromatography, followed by cation and anion exchange chromatography before analysis (see Supplementary material for details).
SAXS data were measured at 23°C using an Anton-Paar SAXSess line collimation instrument (10 mm) equipped with a charge-coupled device detector as previously described (Jeffries et al., 2008). Data were obtained from samples of WT-βTm (3.4 mg/ml), K7del-βTm (3.0 mg/ml), the final dialysate (solvent blank) and water. Details of data reduction are included in the Supplementary material.
Circular dichroism spectra of WT-βTm (1.6 μM) and K7del-βTm (1.4 μM) were recorded on a Jasco J-815 spectropolarimeter. Circular dichroism data were collected at 20°C over the wavelength range 185–260 nm, in a 1.0-mm path length cell, with a resolution of 0.5 nm, a bandwidth of 1 nm and a digital integration time of 1 s. Final spectra were the sum of three scans accumulated at a speed of 20 nm/min and were baseline corrected. Estimates of secondary structure were made using the CDPro suite of programs (Sreerama et al., 2000).
Results
Clinical, magnetic resonance imaging and histological findings
We identified five families from Australia, France (two families from different regions), Germany and Brazil that had the same TPM2 mutation. Detailed clinical information was available on eight affected individuals (Table 1; Fig. 1A–E). The most common presentation was in infancy/early childhood with joint contractures. Three patients had some congenital contractures, and some were mildly delayed in acquiring motor milestones. Most patients were noted to have awkward gaits during childhood, and many required release of tendo-Achilles contractures. Contractures of the jaw, long finger flexors, shoulders and hips (including unusual hip extensor contractures in Patient 3; Fig. 1I) developed during childhood or adulthood in many patients. Patient 2 had prominent pes planus and Patient 8 had marked pes cavus. Patient 2 had moderate generalized muscle hypertonia, most prominent at the limits of muscle extension and in the lower limbs. Proximal lower limb muscle weakness developed in most patients during mid-adulthood, occasionally beginning in adolescence. Some patients also had weakness of the proximal upper limb, ankle dorsiflexor and intrinsic hand muscles. Several patients reported mild dysphagia. Two patients lost ambulation in late adulthood and others had reduced stamina and myalgias (prominent in Patient 3). Patient 8 required non-invasive nocturnal ventilatory support at age 54 years. In Family C, the two affected females were noticeably weaker than their four affected male relatives. All patients had normal cardiac function. All six members of Family C with muscle symptoms (but no other family members) developed psychosis, confirmed as schizophrenia in two individuals. There were no reports of mental illness in other families. Creatine kinase levels were often raised 1.5–4 times the upper limit of normal, and were sometimes markedly elevated. Patient 3 had one possible episode of rhabdomyolysis at age 15 years, associated with red urine.
Clinical and pathological features of K7del patients
| Patient/family/sex/age | Presentation (age) | Clinical features | Contractures | Muscle biopsied (age) | Notable histological features |
|---|---|---|---|---|---|
| 1/A/F/45 | Abnormal gait (early childhood) | Pectus carinatum, mild proximal leg weakness, mild dysphagia, CK 1–1.5× | Jaw, shoulders, hips | Quad (22) | +++ rods, ++ internal nuclei, split fibres, angular atrophic fibres, core-like regions |
| 2/A/M/14 | Delayed walking, awkward gait, falls | Bilateral inguinal hernias, pes planus, in-toe gait, pectus carinatum, thoracic kyphosis, tendo-Achilles releases. Muscle hypertonia. | Ankles, shoulders, hamstrings, jaw. | Quad (7) | +++ rods, +++ core-like regions |
| 3/B/M/41 | Contractures (early childhood) | Marked fatigue and myalgias with exertion, CK 2-20× | Jaw, hips (hip extensors), shoulders, finger flexors ankles, neck extensors | Delt (41) | ++ rods, +++ core-like regions, angular atrophic fibres, + internal nuclei |
| 4/C/F/74 | Contractures (birth). | Proximal limb weakness, severe kyphoscoliosis, ambulation lost by 74 yrs, schizophrenia, CK 2.5× | Congenital: elbows, knees, ankles. Later onset: Jaw, pectoral, finger extensors, quad, hamstrings | Quad (53) | +++ rods, +++ internal nuclei, split fibres, sarcomeric disarray, nuclear clumps |
| 5/C/F/50 | Contractures (birth) | Proximal limb weakness, difficulty walking (50 yrs), schizophrenia | Congenital: elbows, knees, ankles. Later onset: jaw, pectoral, ankle dorsiflexors, quad, hamstrings | Delt (33) | +++ rods, + internal nuclei, increased oxidative staining and myofibril disarray in rod-rich regions |
| 6/C/M/40 | Contractures (birth) | Mild proximal limb weakness, psychosis | Congenital: elbows, knees, ankles. | NA | NA |
| 7/D/M/13 | Abnormal gait (infancy) | Mild proximal limb and distal leg weakness, mild chest deformity and scoliosis, epilepsy | Jaw, ankles | ?site (2) | +++ rods, type 1 fibre predominance |
| 8/E/F/65 | Delayed milestones, toe walking (infancy) | Tendo-Achilles releases, myalgias, quad weakness from 34 yrs, ambulation lost 62 yrs, NCPAP from 54 yrs, CK 1.5–3.5× | Ankles, toes, finger flexors, thumb adductors, jaw | Quad (37) | +++ rods, ++ internal nuclei, split fibres, nuclei clumps, fibre type grouping |
| Patient/family/sex/age | Presentation (age) | Clinical features | Contractures | Muscle biopsied (age) | Notable histological features |
|---|---|---|---|---|---|
| 1/A/F/45 | Abnormal gait (early childhood) | Pectus carinatum, mild proximal leg weakness, mild dysphagia, CK 1–1.5× | Jaw, shoulders, hips | Quad (22) | +++ rods, ++ internal nuclei, split fibres, angular atrophic fibres, core-like regions |
| 2/A/M/14 | Delayed walking, awkward gait, falls | Bilateral inguinal hernias, pes planus, in-toe gait, pectus carinatum, thoracic kyphosis, tendo-Achilles releases. Muscle hypertonia. | Ankles, shoulders, hamstrings, jaw. | Quad (7) | +++ rods, +++ core-like regions |
| 3/B/M/41 | Contractures (early childhood) | Marked fatigue and myalgias with exertion, CK 2-20× | Jaw, hips (hip extensors), shoulders, finger flexors ankles, neck extensors | Delt (41) | ++ rods, +++ core-like regions, angular atrophic fibres, + internal nuclei |
| 4/C/F/74 | Contractures (birth). | Proximal limb weakness, severe kyphoscoliosis, ambulation lost by 74 yrs, schizophrenia, CK 2.5× | Congenital: elbows, knees, ankles. Later onset: Jaw, pectoral, finger extensors, quad, hamstrings | Quad (53) | +++ rods, +++ internal nuclei, split fibres, sarcomeric disarray, nuclear clumps |
| 5/C/F/50 | Contractures (birth) | Proximal limb weakness, difficulty walking (50 yrs), schizophrenia | Congenital: elbows, knees, ankles. Later onset: jaw, pectoral, ankle dorsiflexors, quad, hamstrings | Delt (33) | +++ rods, + internal nuclei, increased oxidative staining and myofibril disarray in rod-rich regions |
| 6/C/M/40 | Contractures (birth) | Mild proximal limb weakness, psychosis | Congenital: elbows, knees, ankles. | NA | NA |
| 7/D/M/13 | Abnormal gait (infancy) | Mild proximal limb and distal leg weakness, mild chest deformity and scoliosis, epilepsy | Jaw, ankles | ?site (2) | +++ rods, type 1 fibre predominance |
| 8/E/F/65 | Delayed milestones, toe walking (infancy) | Tendo-Achilles releases, myalgias, quad weakness from 34 yrs, ambulation lost 62 yrs, NCPAP from 54 yrs, CK 1.5–3.5× | Ankles, toes, finger flexors, thumb adductors, jaw | Quad (37) | +++ rods, ++ internal nuclei, split fibres, nuclei clumps, fibre type grouping |
All ages in years (yrs). CK 2× = serum creatine kinase twice upper limit of normal.
+++ = present in >50% of fibres; ++ = moderately frequent; + = minor abnormality.
Delt = deltoid; F = female; M = male; NA = not applicable; NCPAP = nocturnal continuous positive airway pressure ventilation; Quad = quadriceps.
Clinical and pathological features of K7del patients
| Patient/family/sex/age | Presentation (age) | Clinical features | Contractures | Muscle biopsied (age) | Notable histological features |
|---|---|---|---|---|---|
| 1/A/F/45 | Abnormal gait (early childhood) | Pectus carinatum, mild proximal leg weakness, mild dysphagia, CK 1–1.5× | Jaw, shoulders, hips | Quad (22) | +++ rods, ++ internal nuclei, split fibres, angular atrophic fibres, core-like regions |
| 2/A/M/14 | Delayed walking, awkward gait, falls | Bilateral inguinal hernias, pes planus, in-toe gait, pectus carinatum, thoracic kyphosis, tendo-Achilles releases. Muscle hypertonia. | Ankles, shoulders, hamstrings, jaw. | Quad (7) | +++ rods, +++ core-like regions |
| 3/B/M/41 | Contractures (early childhood) | Marked fatigue and myalgias with exertion, CK 2-20× | Jaw, hips (hip extensors), shoulders, finger flexors ankles, neck extensors | Delt (41) | ++ rods, +++ core-like regions, angular atrophic fibres, + internal nuclei |
| 4/C/F/74 | Contractures (birth). | Proximal limb weakness, severe kyphoscoliosis, ambulation lost by 74 yrs, schizophrenia, CK 2.5× | Congenital: elbows, knees, ankles. Later onset: Jaw, pectoral, finger extensors, quad, hamstrings | Quad (53) | +++ rods, +++ internal nuclei, split fibres, sarcomeric disarray, nuclear clumps |
| 5/C/F/50 | Contractures (birth) | Proximal limb weakness, difficulty walking (50 yrs), schizophrenia | Congenital: elbows, knees, ankles. Later onset: jaw, pectoral, ankle dorsiflexors, quad, hamstrings | Delt (33) | +++ rods, + internal nuclei, increased oxidative staining and myofibril disarray in rod-rich regions |
| 6/C/M/40 | Contractures (birth) | Mild proximal limb weakness, psychosis | Congenital: elbows, knees, ankles. | NA | NA |
| 7/D/M/13 | Abnormal gait (infancy) | Mild proximal limb and distal leg weakness, mild chest deformity and scoliosis, epilepsy | Jaw, ankles | ?site (2) | +++ rods, type 1 fibre predominance |
| 8/E/F/65 | Delayed milestones, toe walking (infancy) | Tendo-Achilles releases, myalgias, quad weakness from 34 yrs, ambulation lost 62 yrs, NCPAP from 54 yrs, CK 1.5–3.5× | Ankles, toes, finger flexors, thumb adductors, jaw | Quad (37) | +++ rods, ++ internal nuclei, split fibres, nuclei clumps, fibre type grouping |
| Patient/family/sex/age | Presentation (age) | Clinical features | Contractures | Muscle biopsied (age) | Notable histological features |
|---|---|---|---|---|---|
| 1/A/F/45 | Abnormal gait (early childhood) | Pectus carinatum, mild proximal leg weakness, mild dysphagia, CK 1–1.5× | Jaw, shoulders, hips | Quad (22) | +++ rods, ++ internal nuclei, split fibres, angular atrophic fibres, core-like regions |
| 2/A/M/14 | Delayed walking, awkward gait, falls | Bilateral inguinal hernias, pes planus, in-toe gait, pectus carinatum, thoracic kyphosis, tendo-Achilles releases. Muscle hypertonia. | Ankles, shoulders, hamstrings, jaw. | Quad (7) | +++ rods, +++ core-like regions |
| 3/B/M/41 | Contractures (early childhood) | Marked fatigue and myalgias with exertion, CK 2-20× | Jaw, hips (hip extensors), shoulders, finger flexors ankles, neck extensors | Delt (41) | ++ rods, +++ core-like regions, angular atrophic fibres, + internal nuclei |
| 4/C/F/74 | Contractures (birth). | Proximal limb weakness, severe kyphoscoliosis, ambulation lost by 74 yrs, schizophrenia, CK 2.5× | Congenital: elbows, knees, ankles. Later onset: Jaw, pectoral, finger extensors, quad, hamstrings | Quad (53) | +++ rods, +++ internal nuclei, split fibres, sarcomeric disarray, nuclear clumps |
| 5/C/F/50 | Contractures (birth) | Proximal limb weakness, difficulty walking (50 yrs), schizophrenia | Congenital: elbows, knees, ankles. Later onset: jaw, pectoral, ankle dorsiflexors, quad, hamstrings | Delt (33) | +++ rods, + internal nuclei, increased oxidative staining and myofibril disarray in rod-rich regions |
| 6/C/M/40 | Contractures (birth) | Mild proximal limb weakness, psychosis | Congenital: elbows, knees, ankles. | NA | NA |
| 7/D/M/13 | Abnormal gait (infancy) | Mild proximal limb and distal leg weakness, mild chest deformity and scoliosis, epilepsy | Jaw, ankles | ?site (2) | +++ rods, type 1 fibre predominance |
| 8/E/F/65 | Delayed milestones, toe walking (infancy) | Tendo-Achilles releases, myalgias, quad weakness from 34 yrs, ambulation lost 62 yrs, NCPAP from 54 yrs, CK 1.5–3.5× | Ankles, toes, finger flexors, thumb adductors, jaw | Quad (37) | +++ rods, ++ internal nuclei, split fibres, nuclei clumps, fibre type grouping |
All ages in years (yrs). CK 2× = serum creatine kinase twice upper limit of normal.
+++ = present in >50% of fibres; ++ = moderately frequent; + = minor abnormality.
Delt = deltoid; F = female; M = male; NA = not applicable; NCPAP = nocturnal continuous positive airway pressure ventilation; Quad = quadriceps.
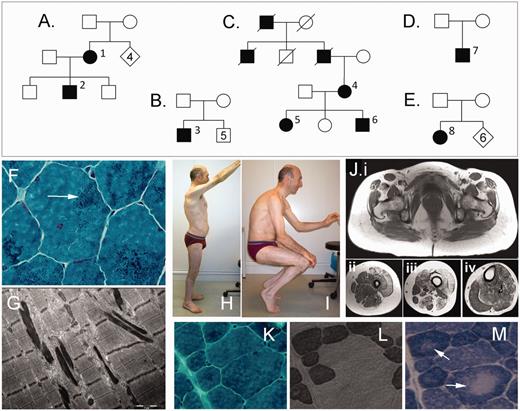
Clinical and histological images and family trees of patients with the p.K7del mutation in TPM2. (A–E) Pedigrees for the respective families. Superscript numerals correspond to individuals in Table 1. (F) Gomori trichrome stain from Patient 2 shows widespread scattered small nemaline bodies (arrow) in most myofibres. (G) Electron microscopy shows typical nemaline bodies as highly electron-dense rods. (H and I) Photographs of Patient 3 show limited ranges of motion of shoulders and hips due to muscle contractures. (J) T1-weighted MRI images of Patient 1 shows fatty infiltration in muscles of the pelvis (Ji), upper thigh (Jii), lower thigh (Jiii) and calf (Jiv). (K–M) equivalent regions of frozen muscle showing that nemaline bodies (K; Gomori trichrome stain) and core-like regions (M; arrows, NADH stain) are present in both type 1 fibres (L; ATPase stain pH 4.3, dark fibres) and type 2 fibres (L; pale fibres) and co-localize poorly.
A lower limb muscle MRI scan of Patient 1, performed at age 47 years, showed mild generalized fatty infiltration of most muscles, with marked involvement of the quadriceps, biceps femoris and semimembranosus muscles (Fig. 1J). Patient 3 had a CT scan at age 42 years that showed fatty infiltration in the quadriceps and medial gastrocnemius muscles.
Muscle biopsies were obtained from seven individuals, mostly in mid-adulthood, and three frozen muscle biopsies were available for further analysis. By light and electron microscopy examination, all biopsies showed numerous small scattered nemaline bodies in >50% of myofibres (Fig. 1F and G), often with subsarcolemmal accumulations. Most biopsies showed marked variation in myofibre size, a tendency to type 1 fibre atrophy and type 1 fibre predominance (>55%) (Supplementary Table 1). Many biopsies showed subsarcolemmal accumulations of mitochondria, and three biopsies showed regions of reduced nicotinamide adenine dinucleotide hydroxyl (NADH) staining in the centre of many myofibres, indicating areas with fewer mitochondria (Fig. 1M). With electron microscopy, we did not identify regions lacking mitochondria, and so the pale regions on oxidative stains likely represent areas with a lower density rather than an absence of mitochondria, and are therefore not typical of true cores. These ‘core-like’ areas were present in both type 1 and type 2 fibres and lacked sharply defined boundaries. We compared sequential sections stained with Gomori trichrome, NADH and ATPase stains and found no correlation between core-like areas and areas rich in nemaline bodies (Fig. 1K–M). Muscle biopsies from older patients showed prominent internal nuclei, myofibre splitting (features that are more often associated with dystrophic processes) and nuclear clumps. Myofibrillar disorganization was common in markedly atrophied fibres and in rod-rich regions in biopsies from older patients.
Genetic analysis
In probands from all five families, we identified the same heterozygous missense three base deletion (c.19_21delAAG) in exon 1 of TPM2. This in-frame deletion is predicted to remove one of three highly conserved lysine residues that occupy amino acid positions 5, 6 and 7 at the N-terminus of WT-βTm (WT-βTm: MDAIKKKMQ … ; K7del-βTm: MDAIKKMQ … ; Fig. 2A). In all families, this mutation tracked with disease and the inheritance pattern in Families A and C are consistent with autosomal dominant inheritance. De novo occurrence in the proband or oldest affected family member was suspected for Families A, B, D and E and confirmed by genetic testing of unaffected parents in Families A and B. The c.19_21delAAG change was not observed in the Exome Variant Server, The National Heart Lung and Blood Institute Exome Sequencing Project (http://evs.gs.washington.edu/EVS/, accessed April, 2012), which contains data from >5000 individuals. Other causes of nemaline myopathy that were excluded by gene sequencing included ACTA1 (all families) and TPM3 (Families A, B and C).
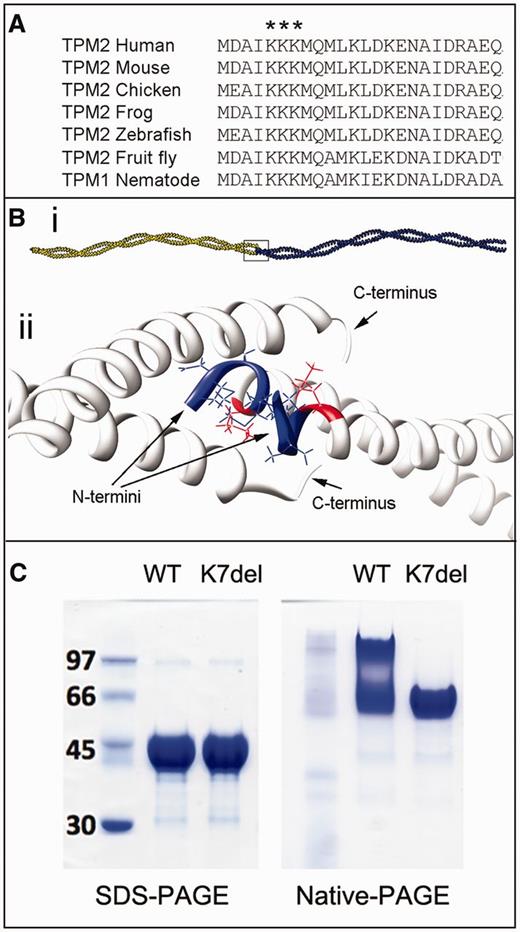
K7del-βTm modelling and polymerization studies. (A) Alignment of the N-terminus of sarcomeric tropomyosin sequences in multiple species demonstrates the high degree of conservation for this region of tropomyosin, including full conservation of K5, K6 and K7. (Bi) Adjacent tropomyosin dimers bind in a head-to-tail manner to form a long polymer. (Bii) Enlargement of box in Bi. Model of the short four-helix bundle at the tropomyosin junction that encompasses the first lysine residue (K5; red) within the N-terminal region of β-tropomyosin (blue). If K7del-βTm molecules dimerize appropriately along central and C-terminal regions, the deletion of an N-terminal lysine (arbitrarily named K7) will manifest as an absence of K5 and misalignment of the first four N-terminal residues (blue). (C) Recombinant WT-βTm runs as both a tropomyosin dimer and higher molecular weight species in native-PAGE gels which likely represent polymerized dimers. In comparison, K7del-βTm appears as a dimer only, suggesting an impaired ability to polymerize.
Molecular modelling predicts abnormal tropomyosin dimerization and Ca2+ regulation
Tropomyosin polymers have a high affinity for actin filaments (Hitchcock-DeGregori, 2008), and the N- and C-terminal overlap region between neighbouring tropomyosin subunits that mediates polymerization also interacts with troponin T and F-actin (Mak et al., 1981a, b; Li et al., 2002). The functional significance of the head-to-tail overlap region with respect to function is highlighted by the observation that bacterially expressed recombinant α-tropomyosins, which lack N-terminal acetylation of the first methionine residue, do not end-on-end associate and are functionally perturbed (Monteiro et al., 1994). To predict the possible effects of the p.K7del mutations on tropomyosin function, we used a molecular model based on the crystal structure of α-tropomyosinfast (Whitby et al., 2000) and the structures of α-tropomyosin peptides encompassing the head-to-tail overlap region (Greenfield et al., 2006; Frye et al., 2010; Li et al., 2011). The ∼10 N- and C-terminal amino acids of neighbouring tropomyosin dimers interact to create a short four-helix bundle head-to-tail junction (Fig. 2B). The fact that the p.K7del mutation is located within this key overlap region suggests that the removal of K7 may disrupt β-tropomyosin polymerization and/or affect interactions with actin or troponin T.
Biophysical analyses of K7del structure
A qualitative assessment of the SAXS data derived from overlaying the scattering profiles of WT-βTm and K7del-βTm [I(q) versus q] suggest that the mutation does not grossly perturb the overall shape of tropomyosin in solution. Both SAXS profiles overlay reasonably well and log[I(q)] versus q plots and subsequent Kratky analyses [I(q)q2 versus q] have characteristic features indicative of extended particles that possess a degree of structural flexibility (Supplementary Fig. 4A and Supplementary Data). The indirect Fourier transformation of I(q) versus q yields the atom-pair distribution function for the scattering [P(r) versus r], and a comparison of P(r) for WT-βTm and K7del-βTm shows that both proteins have highly extended rod-like shapes that are similar to the P(r) calculated from the α-tropomyosinfast crystal structure (Supplementary Fig. 4B). Estimates of the molecular mass of the scattering particles from the forward scattering [I(0)] indicate that both the WT-βTm and K7del-βTm molecules are mostly in dimers in solution (Supplementary Table 2). However, it must be noted that the maxima in the experimentally derived P(r)s for both constructs occur at a distance that is slightly smaller compared with that for the crystal structure (17–20 Å compared with ∼22 Å), and the secondary maxima have been significantly smoothed and possibly shifted, suggesting that a small portion of monomers with a smaller cross-sectional diameter are present in solution for both samples. The circular dichroism results indicate that K7del-βTm and WT-βTm are folded in solution and are predominantly α-helical (Supplementary Fig. 5).
Reducing SDS-PAGE analysis reveals that WT-βTm and K7del-βTm migrate to the same extent on the gel under denaturing conditions (Fig. 2C). However, when SDS is omitted and the proteins are run in native reducing conditions, the WT-βTm migrates as a smear of mixed species with apparent increasing mass-to-charge ratios, which likely represent polymerized dimers. In contrast, K7del-βTm continues to migrate as a single band (i.e. as a dimer), suggesting an impaired ability to polymerize.
Normal tropomyosin isoform ratios in K7del patient muscle
The nemaline myopathy p.M9R mutation in TPM3 is associated with abnormal tropomyosin ratios in patient muscle (Corbett et al., 2005). Levels of β-tropomyosin in muscle from two K7del patients were normal compared with age- and fibre-type matched controls (Supplementary Fig. 1), which exclude abnormal tropomyosin isoform ratios as a disease mechanism in this myopathy.
Evidence of apoptosis and mitochondrial dysregulation
To investigate whether nuclear clumps may reflect increased apoptosis, we used a TUNEL assay to detect nuclei undergoing DNA fragmentation. Levels of TUNEL-positive nuclei in Patient 3 were raised (0.625%) compared with muscle from four healthy control subjects (0.067–0.394) (Supplementary Fig. 1). Western blotting for porin and mitochondrial enzyme complexes I–V showed that K7del patients had more variable protein levels than four healthy control subjects, providing evidence for mitochondrial dysregulation (Supplementary Fig. 2).
Mutant K7del-β-tropomyosin protein incorporates poorly into sarcomeres and likely accumulates in nemaline bodies
The deletion of a basic N-terminal lysine alters the predicted isoelectric point of β-tropomyosin from 4.66 to 4.64, which permits the separation of K7del-βTm and WT-βTm by 2D-PAGE. To determine whether mutant protein is expressed in patient muscle, we performed 2D-PAGE on the filamentous fraction of muscle lysates, which represents tropomyosin bound in high molecular weight complexes such as sarcomeres and nemaline rods. An extra spot consistent with K7del-βTm was seen in all three available biopsies (Fig. 3Aii–iv, white arrow) at levels higher than WT-βTm (Fig. 3Aii-iv, black arrow).
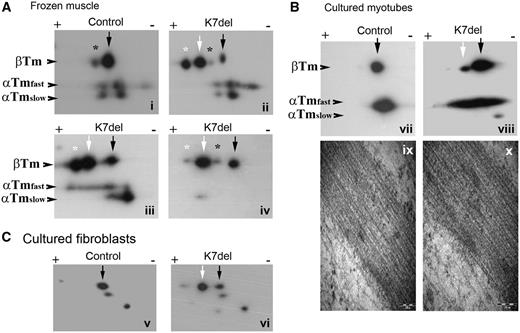
2D-PAGE analysis of K7del-βTm and WT-βTm. The K7del mutation results in the loss of a basic lysine residue allowing the separation of the WT and mutant βTm protein by 2D-PAGE. K7del-βTm (Aii, white arrow) has a more acidic isoelectric point (4.64) than WT-βTm (4.66); (Ai, black arrow). (Ai–iv) In K7del patient muscle, K7del-βTm (white arrow) is more abundant than WT-βTm (black arrow). All sarcomeric tropomyosin isoforms have phosphorylated protein species (asterisk), especially in muscle from young children (Ai). K7del-βTm is highly phosphorylated (white asterisk) that WT-βTm (black asterisk; Aii and iii). Lambda phosphatase treatment of muscle homogenates markedly reduces phosphorylated K7del-βTm (Aiv). Panel B shows tropomyosin levels in phosphatase-treated primary cultured myotubes (insoluble protein fraction only to assess protein bound in high molecular weight complexes). In this system, K7del-βTm levels (white arrow) are much lower than WT-βTm (black arrow) in K7del-βTm myotubes suggesting K7del-βTm incorporates poorly into developing sarcomeres. No abnormal protein inclusions such as nemaline rods are seen in control (Bix) or K7del primary cultured myotubes (Bx) on electron microscopy. Panel C shows the cytoskeletal tropomyosin isoforms present in cultured primary human fibroblasts. In addition to normal TM1 (black arrows), a more acidic protein species, consistent with K7del-TM1 (white arrow) is present in K7del fibroblasts but not control cells. In panels A and B, staining with the TPM311 antibody shows sarcomeric tropomyosin isoforms. In panel C, staining with the γ9d antibody shows cytoskeletal tropomyosin isoforms.
The amount of mutant protein that incorporates into sarcomeres is likely to be a key factor that determines disease severity. The presence of nemaline rods in patient muscle, which sediment with centrifugation together with sarcomeric proteins, makes it difficult to quantify the levels of mutant protein within sarcomeres alone. To overcome this difficulty, we studied patient primary cultured myotubes that lack nemaline bodies based on phalloidin staining and electron microscopy studies (Fig. 3Bix and x) similarly to wild type myotubes (Fig. 3Bix). We confirmed that these cells express both K7del-βTm and WT-βTm messenger RNA species by complementary DNA analysis (data not shown). We performed 2D-PAGE on cultured myotubes differentiated to Day 11, which contain well-formed striations (data not shown). In the filamentous protein fraction, the level of K7del-βTm (Fig. 3Bviii, white arrow) was much lower than WT-βTm protein (Fig. 3Bviii, black arrow), which is the inverse of the result from patient muscle containing nemaline bodies. These data suggest that little K7del-βTm incorporates into sarcomeres, compared with WT-βTm. The most likely explanation for the abundance of mutant K7del-βTm in the filamentous protein fraction in patient muscle is that the majority resides in nemaline bodies.
Impaired incorporation of K7del-β-tropomyosin into sarcomeres in cell culture
We examined the ability of β-tropomyosin constructs tagged with enhanced green fluorescent protein (βTm-EGFP) to incorporate into sarcomeres in differentiating C2C12 myotubes. Cells transfected with K7del-βTm-EGFP or WT-βTm-EGFP were harvested 24 h after transfection (Day 0) and after 3 days of differentiation (Day 3). Soluble and insoluble protein fractions were extracted by low spin centrifugation (13 000g). There was a significant decrease in the proportion of β-tropomyosin in the insoluble protein pool after myotube differentiation, suggesting that K7del-βTm incorporates less well into sarcomeres than WT-βTm (Fig. 4Bi and ii)). No significant difference was seen in recently transfected (Day 0) myoblasts, indicating that K7del-βTm incorporates into the actin cytoskeleton although stress fibres appeared less abundant (Fig. 4Aii) as compared to WT-βTm (Fig. 4Ai), suggesting that mutant K7del-βTm impairs stress fibre formation.
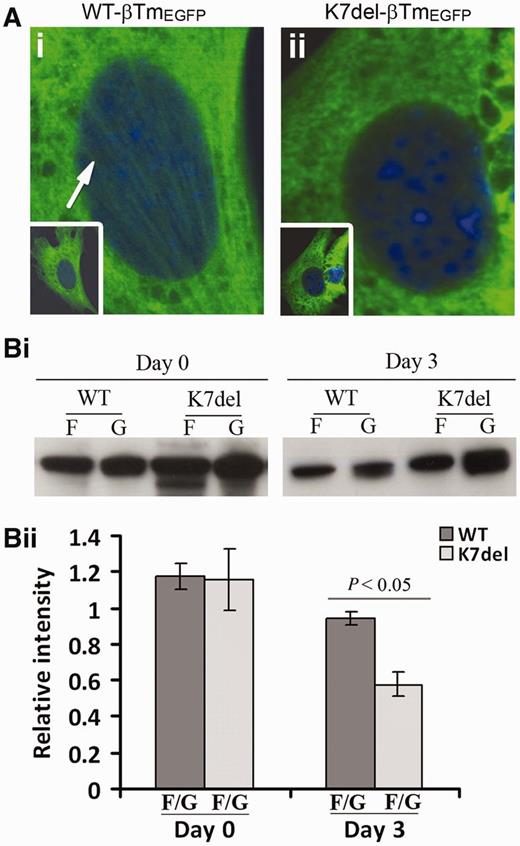
K7del-βTm incorporates poorly into sarcomeres in differentiated C2C12 cells. (A) Representative undifferentiated (Day 0, D0) C2C12 cells transfected with constructs encoding either (i) WT-βTm tagged with enhanced green fluorescent protein (EGFP) or (ii) K7del-βTm similarly tagged. Actin stress fibre labelling with WT-βTm-EGFP was more prominent than with K7del-βTm-EGFP. (Bi) Representative western blot images showing levels of βTm-EGFP in insoluble protein pools 0 and 3 days after differentiation. (ii) In triplicate experiments, significantly less β-tropomyosin-EGFP was present in the insoluble (F) protein fraction (representing protein bound in high molecular weight complexes) for K7del-βTm compared with WT-βTm constructs, consistent with impaired incorporation of K7del-βTm into developing sarcomeres. G = soluble fraction.
K7del-β-tropomyosin patient myofibres generate normal force and have increased myofilament Ca2+ sensitivity
Results for both maximum force and myofilament Ca2+ sensitivity measurements were similar in myofibres from two unrelated K7del patients, and results were pooled. Maximal specific force generation was not significantly different in patient and control myofibres (Fig. 5A). The myofilament Ca2+ sensitivity was consistently increased in both type 1 and type 2A patient myofibres, indicated by leftward shifts in the pCa-force curves in Fig. 5Bi and Bii, with the most marked increase in type 1 fibres. The pCa50 (i.e. the Ca2+ concentration required to generate 50% of maximal force) was 6.76 ± 0.03 in type 1 K7del patient myofibres compared with 6.32 ± 0.14 in controls (P < 0.001, Fig. 5Bi), and the pCa50 was 6.36 ± 0.01 in patient type 2A myofibres compared with 6.13 ± 0.04 in controls (P < 0.001, Fig. 5Bii).
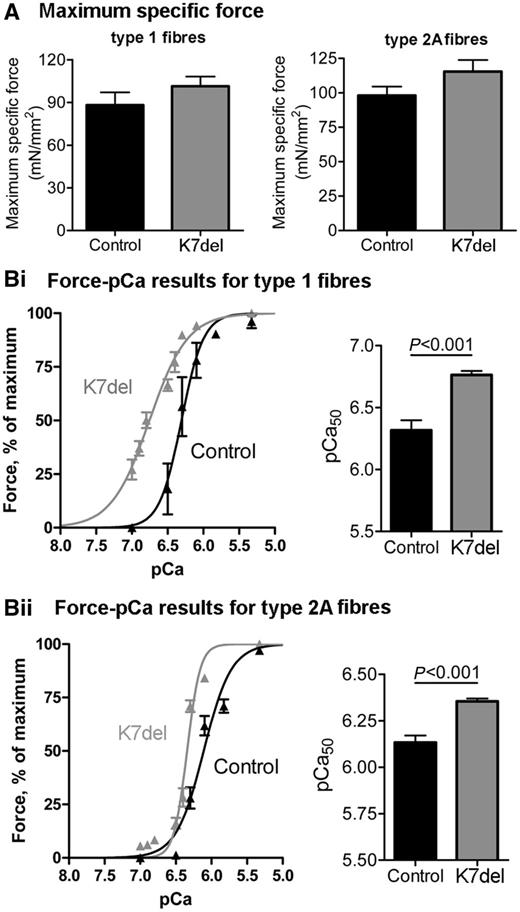
Normal force generation and increased myofibrillar Ca2+ sensitivity in K7del patient myofibres. (A) Maximum specific force was not statistically different between controls and K7del patients in either type 1 or type 2A myofibres. (Bi and ii) The Ca2+ sensitivity of muscle contraction was increased in both type 1 and type 2A myofibres from K7del patients compared with control myofibres.
In vitro motility assays also show increased Ca2+ sensitivity in K7del patient myofibres
When 100% recombinant K7del-βTm protein was incorporated into thin filaments in the in vitro motility assay, it was difficult to obtain Ca2+-sensitive regulation of filament motility, even when high concentrations of K7del-βTm and troponin were used (data not shown). In patient muscles, K7del-βTm is co-expressed with WT-βTm (Fig. 3). When we mimicked this situation by mixing 50% mutant and 50% wild-type protein constructs in vitro, we obtained good Ca2+ regulation of filament movement with 50 nM tropomyosin, indicating that it was functionally incorporated into the thin filament. Ca2+ sensitivity was consistently higher when 50% K7del-βTm was used than with WT-βTm alone (K7del + WT/WT EC50 ratio 0.45 ± 0.25), and sliding speed at fully activating Ca2+ concentrations was higher (ratio of K7del + WT/WT filament velocity = 1.8 ± 0.9; Fig. 6).
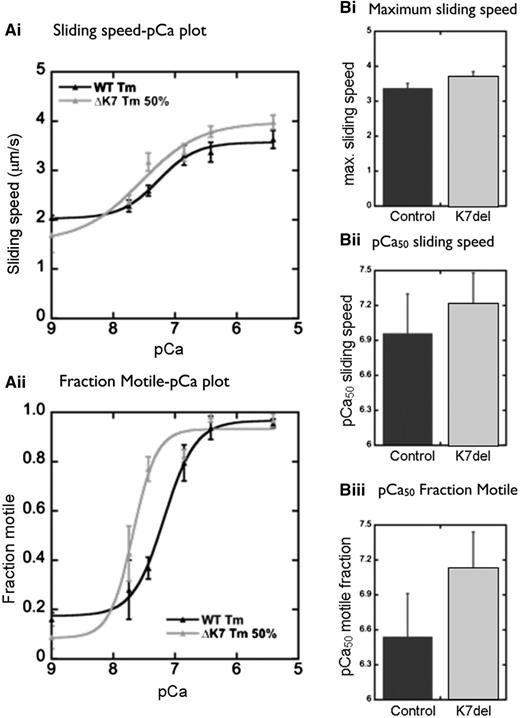
In vitro motility assay results. A 50/50 mixture of WT-βTm and K7del-βTm (ΔK7) confers an increase in filament velocity at a range of activating Ca2+ concentrations (Ai), an increase in maximum sliding speed (Bi) and increased sensitivity of actin filaments to Ca2+ (Aii, Bii and iii) compared with control (100% WT-βTm).
K7del-β-tropomyosin has reduced binding affinity for actin filaments
In the actin-tropomyosin co-sedimentation assay, WT-βTm behaved as expected showing extrapolated maximum binding at 4.0 µM, consistent with tropomyosin saturating actin at 0.22 mol tropomyosin/mol actin (molecular ratio of ∼1:9, close to the expected ratio of 1:7). Actin-binding affinity was around 6-fold lower for K7del-βTm compared with WT-βTm, and maximum binding was not achieved using our assay conditions (Supplementary Data and Supplementary Fig. 3). Based on the co-sedimentation data, the approximate disassociation constant (Kd) and Hill coefficient between WT-βTm and F-actin was measured at 6.1 μM and 3.7, respectively, while the mutation severely limited actin binding (Kd ∼ 37 μM), making it difficult to ascertain whether any cooperativity was present in the interaction between K7del-βTm and actin (h ∼ 1.3) .
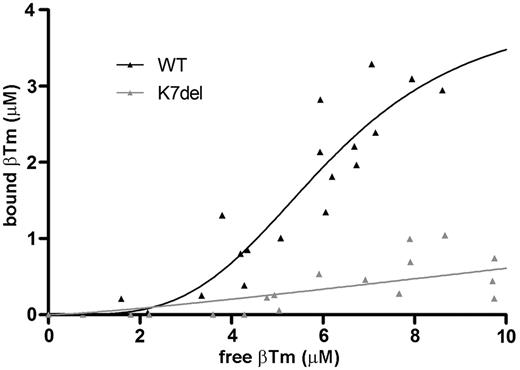
Actin-βTm co-sedimentation.
Discussion
This study suggests that the p.K7del TPM2 mutation is responsible for a substantial proportion of disease associated with this gene, perhaps ∼25% of families. The five families we report are almost certainly unrelated and each likely represents a de novo occurrence of the p.K7del mutation (confirmed in two families). The likely reason the p.K7del mutation recurs is that short repetitive genetic sequences (in this case a sequence of AAG codons that encode three sequential lysine residues) can act as deletion hotspots because slipped-strand mispairing can occur when DNA is copied or repaired, leading to deletion of a repeat. This mechanism is accepted as the basis for other recurrent deletions and also likely accounts for the one other recurrent TPM2 mutation reported to date, p.E132del (Kearney et al., 2006; Lehtokari et al., 2007; Clarke et al., 2009). The probable reason that p.K7del has not been reported in TPM3, which is fully homologous at the protein level for this region, is that lysine 6 is encoded by an AAA codon in TPM3, which breaks the repeat sequence.
All patients with the p.K7del mutation in TPM2 shared an unusual pattern of clinical features. Most congenital myopathies present with muscle weakness and hypotonia in early life, while K7del-TPM2 patients usually presented in infancy with joint contractures and abnormal gait, and multiple joint contractures remained the most prominent clinical problem until mid-life. In light of the study findings, we re-examined Patient 2 and found clear generalized muscle hypertonia, an unusual finding in congenital myopathies, which was missed on previous assessments. Mutations in TPM2 have been previously associated with contractures, most often distal arthrogryposis with prominent contractures of the hands or feet at birth (Sung et al., 2003; Tajsharghi et al., 2007). The phenotype associated with the p.K7del mutation differs in that contractures were only sometimes present at birth, and most developed post-natally and were slowly progressive. In three of seven muscle biopsies, core-like regions that represent mitochondria-scarce regions were also found in numerous myofibres, and several patients were diagnosed with core-rod myopathy instead of classical nemaline myopathy. These features combine to give a distinctive clinical and histological phenotype that may alert the clinician to a mutation in TPM2.
There is a strong association of the p.K7del mutation and psychosis in Family C but not in other families. Gene splicing patterns predict expression of the p.K7del TPM2 mutation in the TM1 cytoskeletal tropomyosin isoform (Schevzov et al., 2005), which is also transcribed from TPM2, and we found strong evidence for expression of p.K7del-TM1 in fibroblasts from two unrelated patients. Non-muscle phenotypes have not been previously reported with TPM2 mutations but there is evidence that the TM1 isoform is expressed at low levels in brain (Schevzov et al., 2005). The strong association between the p.K7del TPM2 mutation in one family raises the possibility that TPM2 may influence susceptibility to psychosis, or this may be a chance association.
In this study, we combined detailed clinical and histological evaluations with a range of in vitro studies of muscle and protein function to understand the main mechanisms of disease, and to inform the development of specific therapies for p.K7del TPM2 nemaline myopathy and closely related conditions. 2D-PAGE analysis provided strong evidence that K7del-β-tropomyosin is present at high levels in patient muscle and disease almost certainly arises because mutant protein interferes with the function of WT-βTm and/or other cellular processes (i.e. a dominant negative disease mechanism). Much of this study has focused on understanding the fate of K7del-βTm protein, whether it is incorporated into sarcomeric structures or not, and how various aspects of muscle physiology are affected.
2D-PAGE experiments using frozen patient muscle biopsies demonstrated that levels of mutant K7del-βTm were greater than WT-βTm in patient muscle, and using a centrifugation protocol we confirmed that much was bound in high molecular weight complexes. To investigate whether K7del-βTm incorporates into sarcomeres or largely resides in nemaline rods, we studied primary cultured patient myotubes, a rod-free system that expresses both K7del-βTm and WT-βTm species. In this system, mutant K7del-βTm only accounted for a small percentage of β-tropomyosin in high-molecular weight structures, which mostly represents protein within sarcomeres. This result suggests that mutant K7del-βTm incorporates poorly into sarcomeres compared with WT-βTm. Other data support this conclusion. EGFP-tagged K7del-βTm incorporated significantly less well into high molecular weight protein complexes in differentiating C2C12 cells compared with EGFP-tagged WT-βTm. In addition, when recombinant K7del-βTm was added to actin filaments in the in vitro motility assay, we could not reliably obtain Ca2+-regulated actin motility, consistent with a difficulty in achieving K7del-βTm-decorated actin filaments. When we measured the affinity of K7del-βTm for actin filaments in a pull-down assay, we found a 6-fold reduction in binding affinity. These data all indicate that relatively little K7del-βTm is incorporated into sarcomeres, likely because mutant K7del-βTm has a markedly reduced affinity for sarcomeric actin filaments.
Tropomyosin molecules must assemble into dimers and then long polymers for normal function within the sarcomere. Molecular modelling predicted that the p.K7del mutation is unlikely to prevent dimerization, and both SAXS and circular dichroism studies using recombinant β-tropomyosin protein confirmed that K7del-βTm forms dimers with similar biophysical properties to WT-βTm. The N-terminal lysine residues take part in the head-to-tail bond between adjacent tropomyosin molecules and therefore the p.K7del mutation is predicted to affect the ability of β-tropomyosin to polymerize into long filaments. In native PAGE experiments, the absence of high molecular weight species for K7del-βTm, representing polymerized tropomyosin dimers in K7del solution, is consistent with a reduced ability to form polymers, compared with WT-βTm. In addition, a lower Hill coefficient in tropomyosin-actin binding experiments is consistent with reduced cooperativity of K7del-βTm-actin binding, a property that is strongly linked to the ability of tropomyosin to self-polymerize (Mak et al., 1981a).
If mutant K7del-βTm binds filamentous actin with such low affinity in vitro, how can it incorporate into the sarcomere even in low amounts? The answer likely stems from the fact that wild-type isoforms of β-tropomyosin, α-tropomyosinslow and α-tropomyosinfast are also available as dimer partners for mutant K7del-βTm in patient muscle. While the pull-down experiments demonstrated that homodimers of mutant K7del-βTm have a low affinity for filamentous actin, the actin affinity of mutant K7del-βTm/WT-Tm heterodimer is likely to be less abnormal. Indeed, a 50:50 mixture of wild-type and K7del-βTm was able to give reliable, although abnormal, Ca2+ regulation of thin filaments in the in vitro motility assay. We hypothesize that most of the mutant K7del-βTm bound to actin filaments will be present in K7del-βTm/WT-Tm heterodimers.
There is increasing evidence that the mechanism for muscle dysfunction in tropomyosin myopathies depends on the specific mutation. We excluded marked alteration of tropomyosin isoform ratios in K7del myopathies, a disease mechanism associated with the TPM3 p.M9R mutation. Some mutations, such as p.K49del, p.E139del and p.Q147P, impair actin-binding and disruption to the α-helical structure of tropomyosin, and abnormal secondary protein structure likely contribute (Marttila et al., 2012). Other mutations have been associated with abnormal myofilament Ca2+ sensitivity; either increased Ca2+ sensitivity (p.R91G; associated with congenital contractures) or reduced sensitivity (p.E41K, p.E117K) (Robinson et al., 2007; Ochala et al., 2008; Marttila et al., 2012). In myofibres isolated from two frozen biopsies from two unrelated patients with the p.K7del TPM2 mutation, we found a significant increase in the Ca2+ sensitivity of force generation and no reduction in maximum specific force. These data indicate that the K7del mutation alters either β-tropomyosin–troponin interactions, tropomyosin–actin interactions or both. An increase in the myofilament Ca2+ sensitivity indicates that muscle contraction will be activated by lower cytoplasmic Ca2+ levels than usual and therefore increased basal muscle tone is an expected outcome. We found clear hypertonia when we re-examined a 14-year-old boy with the K7del mutation for this feature. The progressive contractures associated with the p.K7del mutation may result from impaired muscle relaxation and a tendency for muscles to slowly shorten. Other sarcomeric mutations associated with contractural phenotypes have been associated with increased Ca2+ sensitivity, although the previously observed pattern is for contractures to arise in utero (Sung et al., 2003; Jain et al., 2012).
Our results suggest that the presence of mutant K7del-βTm does not directly cause muscle weakness in the sarcomere by impairing actin–myosin interactions. This likely explains why most patients do not have clear muscle weakness in childhood. However, all patients that we report developed significant disability from mid-life, such as loss of ambulation, particularly in females. The various biophysical analyses we performed do not account for this later deterioration but clinical investigations offer possible answers. The presence of type 1 fibre predominance and atrophy in most biopsies, and core-like regions and dysregulation of mitochondrial proteins in some may indicate muscle responding to cellular or physiological stress. Biopsies taken at older ages showed disorganization of myofibres, nuclear clumps in atrophic fibres and increased rates of internal nuclei. In addition, muscle MRI scans showed marked fatty infiltration of lower leg muscles. These features indicate an impaired ability to repair and maintain normal muscle structure during adulthood in K7del TPM2 patients, perhaps as a result of chronic cellular stress. The presence of nuclear clumps and TUNEL-positive nuclei strongly suggests that muscle apoptosis is upregulated in K7del TPM2 patients, as was reported in a transgenic mouse model of the p.Met9Arg TPM3 nemaline myopathy mutation (Sanoudou et al., 2003). An overall reduction in functional sarcomeres due to fibre atrophy and sarcomeric disorganization, and/or apoptosis with fatty replacement of myofibres may account for the progressive muscle weakness in middle age.
How do these findings inform the long-term aim of developing therapies for these conditions? Firstly, these data indicate that some congenital myopathy mutations do not directly impair force production in the sarcomere and may, like p.K7del, predispose to increased muscle contraction. Ca2+ sensitizers, a class of drugs that were developed for cardiomyopathies, have been proposed as treatments for skeletal muscle disorders (Ochala, 2010; Ochala et al., 2012). Treatment with Ca2+ sensitizers, which increase the sensitivity of the troponin complex to Ca2+ (Kass et al., 2006), is unlikely to restore muscle strength in patients with the p.K7del TPM2 mutation because Ca2+ sensitivity is already raised, and may accelerate the development of muscle contractures. We recommend caution in the widespread use of Ca2+ sensitizers in congenital myopathies without careful consideration of the pathophysiology underlying each mutation.
A second important conclusion from this study is that the basis for muscle dysfunction in many patients with nemaline myopathy and similar myopathies may not be from direct effects of the mutant protein on muscle contraction in the sarcomere, but from indirect effects on a range of myofibre systems such as growth, maintenance of sarcomeric structure and energy metabolism. As previously discussed, there is indirect evidence that myofibres expressing the p.K7del TPM2 mutation are subject to chronic cellular stress. The main sources of stress are uncertain, but may include a high demand on pathways that manage misfolded proteins such as protein chaperones and proteosomes, autophagy pathways and processes that remodel sarcomeres, such as the calpain proteolytic system (for reviews see Beckmann et al., 2008; Murton et al., 2008; Knaevelsrud et al., 2010). Further research is needed to investigate these possible disease mechanisms. Therapies that target these pathways may be more effective in protecting patients from declining muscle strength with age, a common feature in most congenital myopathies and muscular dystrophies.
Funding
National Health and Medical Research Council of Australia [571287 (to N.C.), 403941 (to K.N.N.), 1022707 (to K.N.N. and N.C.) and 1002147 (to N.L.)], a Foundation Building Strength for Nemaline Myopathy grant (2009; to B.I., K.N.N. and N.C.) a VENI grant from the Dutch Organization for Scientific Research, the seventh Framework Program of the European Union [project “NEMMYOP” (to C.O.)], the Australian Academy of Science (to G.R.), Australian Research Council Future Fellowship [FT100100734 (to K.J.N.)] and an Endeavour international postgraduate research scholarship (EIPRS) from the University of Sydney (to N.M.).
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
The authors thank all patients and family members who provided samples and clinical information for our study and Philippa Stokes for her contributions to circular dichroism experiments.
Abbreviations
- CCD
charge-coupled device
- CD
circular dichroism
- DTT
dithiothreitol
- EGFP
enhanced green fluorescent protein
- EGTA
ethylene glycol-bis tetraacetic acid
- IPG
immobilised pH gradient
- K7del-βTm
K7del-β-tropomyosin recombinant protein
- MES
morpholinoethanesulfonic acid
- NADH-TR
nicotinamide adenine dinucleotide hydroxyl tetrazolium reductase
- NHLBI
The National Heart Lung and Blood Institute
- SAXS
small-angle X-ray scattering
- WT-βTm
wild-type β-tropomyosin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}