




Abstract
Blood transfusion has been identified as a source of human-to-human transmission of variant Creutzfeldt–Jakob disease. Three cases of variant Creutzfeldt–Jakob disease have been identified following red cell transfusions from donors who subsequently developed variant Creutzfeldt–Jakob disease and an asymptomatic red cell transfusion recipient, who did not die of variant Creutzfeldt–Jakob disease, has been identified with prion protein deposition in the spleen and a lymph node, but not the brain. This individual was heterozygous (MV) at codon 129 of the prion protein gene (PRNP), whereas all previous definite and probable cases of variant Creutzfeldt–Jakob disease have been methionine homozygotes (MM). A critical question for public health is whether the prion protein deposition reported in peripheral tissues from this MV individual correlates with infectivity. Additionally it is important to establish whether the PRNP codon 129 genotype has influenced the transmission characteristics of the infectious agent. Brain and spleen from the MV blood recipient were inoculated into murine strains that have consistently demonstrated transmission of the variant Creutzfeldt–Jakob disease agent. Mice were assessed for clinical and pathological signs of disease and transmission data were compared with other transmission studies in variant Creutzfeldt–Jakob disease, including those on the spleen and brain of the donor to the index case. Transmission of variant Creutzfeldt–Jakob disease was observed from the MV blood recipient spleen, but not from the brain, whereas there was transmission from both spleen and brain tissues from the red blood cell donor. Longer incubation times were observed for the blood donor spleen inoculum compared with the blood donor brain inoculum, suggesting lower titres of infectivity in the spleen. The distribution of vacuolar pathology and abnormal prion protein in infected mice were similar following inoculation with both donor and recipient spleen homogenates, providing initial evidence of similar transmission properties after propagation in PRNP codon 129 MV and MM individuals. These studies demonstrate that spleen tissue from a PRNP MV genotype individual can propagate the variant Creutzfeldt–Jakob disease agent and that the infectious agent can be present in the spleen without CNS involvement.
Introduction
Creutzfeldt–Jakob disease is a rare human prion disease that occurs sporadically, in association with inherited gene mutations or may be acquired (Prusiner, 1998). A novel form of Creutzfeldt–Jakob disease, variant Creutzfeldt–Jakob disease, was first reported in the UK in 1996 (Will et al., 1996) and since then 176 UK patients and 51 patients from 11 other countries (www.cjd.ed.ac.uk) have been identified. The hypothesis that variant Creutzfeldt–Jakob disease is caused by infection with bovine spongiform encephalopathy has been supported by a range of evidence, including experimental animal studies demonstrating close similarities between the agent strain of variant Creutzfeldt–Jakob disease and bovine spongiform encephalopathy (Bruce et al., 1997; Hill et al., 1997). Surveillance systems are in place in many countries to monitor both bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease and, while the number of cases of bovine spongiform encephalopathy worldwide is clearly in decline, there is still considerable uncertainty surrounding the future course of the variant Creutzfeldt–Jakob disease outbreaks in the UK and other countries.
The codon 129 polymorphism in the prion protein gene (PRNP) is known to influence susceptibility to Creutzfeldt–Jakob disease (Pocchiari et al., 2004). All definite and probable cases of variant Creutzfeldt–Jakob disease with data on genotype have occurred in individuals homozygous for methionine at codon 129 (MM129). Experimental transmission studies indicate that individuals of all codon 129 genotypes may be susceptible to variant Creutzfeldt–Jakob disease infection (Bishop et al., 2006). The incubation period in individuals with the MV or VV genotypes is likely to be longer than those with the MM genotype and these individuals may never develop clinical disease. This is of major significance to public health as subclinical infection has the potential to increase the risks of onward secondary transmission of infection from person to person, for example by blood transfusion.
The number of individuals with asymptomatic variant Creutzfeldt–Jakob disease infection in the UK population is unknown, with estimates ranging from 900 (statistical modelling; Clarke et al., 2007) to 3800 (screening of anonymous appendix and tonsil tissue archives; Hilton et al., 2004). One recent modelling study, which includes the potential for all codon 129 genotype individuals to be at risk and the possibility of unidentifiable blood transfusion infections, suggests that there will be between 84 and 3000 cases of variant Creutzfeldt–Jakob disease in the years 2010 to 2179 (Garske and Ghani, 2010). The most recent appendix tissue archive survey has indicated a prevalence of asymptomatic variant Creutzfeldt–Jakob disease infection of around 1 in 2000 in the UK population (http://www.hpa.org.uk/hpr/archives/2012/hpr3212.pdf) (HPA, 2012).
The identification in 2004 of the first case of transfusion associated variant Creutzfeldt–Jakob disease infection (Llewelyn et al., 2004), through non-leucodepleted red cells, provided evidence of human-to-human transmission of variant Creutzfeldt–Jakob disease. Two additional cases of transfusion transmission of variant Creutzfeldt–Jakob disease have since been identified in MM individuals (Wroe et al., 2006). In addition a patient, who died of non-Creutzfeldt–Jakob disease related causes, has been shown to have abnormal prion protein deposition in the spleen and a lymph node, but not the brain (Peden et al., 2004). This individual, with an MV genotype, had received a red blood cell transfusion from an MM donor who subsequently died of variant Creutzfeldt–Jakob disease. This is the first direct evidence demonstrating that variant Creutzfeldt–Jakob disease infection can occur in an individual with an MV genetic background.
This is of significant potential importance for public health as ∼50% of Caucasian populations are codon 129 heterozygotes and carrier states of prion infection would increase the risk of secondary transmission of infection. It is therefore of critical importance to determine whether deposition of the disease associated (‘scrapie’) form of the prion protein (PrPSc) in the MV spleen is associated with actual prion infectivity (Wadsworth et al., 2011). Furthermore, there is the potential for the properties of the infectious agent to change after secondary transmission within a species or in a different genetic background and it is therefore important to determine whether the disease agent characteristics in the spleen of the MV individual are distinct from that of primary variant Creutzfeldt–Jakob disease infection in an MM background.
Materials and methods
Mouse lines
Transgenic mice expressing human prion protein (HuMM) have been developed by the Neurobiology Division (The Roslin Institute) by a methodology that is detailed elsewhere (Bishop et al., 2006). These inbred mice are homozygous for methionine at codon 129 and express human prion protein under the control of the original mouse prion gene regulatory region. Wild-type RIII (also referred to as MR) mouse lines are used routinely at The Roslin Institute for transmissible spongiform encephalopathy transmission studies. They have previously been inoculated with a number of isolates from variant Creutzfeldt–Jakob disease and develop clinical disease and pathology at ∼300–450 days post injection dependent on the inoculation material and method (Bruce et al., 2004; Ritchie et al., 2009; Diack et al., 2012).
Inoculation
The variant Creutzfeldt–Jakob disease brain and spleen tissue samples were supplied by the tissue bank in the National Creutzfeldt–Jakob Disease Research and Surveillance Unit, part of the Edinburgh Brain Bank funded by the MRC (G1100616). Consent was given for use of these tissues in research, and the tissue bank has ethics approval (Lothian NHS Board, Research Ethics Committee, Reference: 11/ES/0022). Transgenic mice were anaesthetized with halothane and injected with 0.02 ml of 1% w/v brain or spleen homogenate into the right cerebral hemisphere. Wild-type mice received a 0.02 ml dose of 10% w/v homogenate and an additional intraperitoneal injection of 0.1 ml. From 100 days the mice were scored for signs of disease (Fraser and Dickinson, 1968). Mice were sacrificed by cervical dislocation at the terminal stage of disease or for welfare reasons. The brains of the mice were recovered at post-mortem and cut sagittally, with one half snap-frozen in liquid nitrogen for biochemical analysis and the remaining half fixed for histology. The animal experiments were approved by The Roslin Institute’s ethical review committee and they were conducted according to the regulations of the United Kingdom Home Office Animals (Scientific Procedures) Act 1986.
Prion protein detection by immunocytochemistry
Following fixation in 10% formal saline for 48 h, the mouse half-brains were treated for 1.5 h in 98% formic acid, cut transversely into four sections, and embedded in paraffin. Sections of 6 -µm thickness were cut for immunocytochemistry. Prion protein was detected by use of the Vectastain Elite ABC Kit (Vector Labs) with overnight anti-prion protein primary antibody incubation (6H4, Prionics, 1:5000). Detection of antibody binding was through deposition of 3,3’-diaminobenzidine chromogen.
Scoring of vacuolation
Haematoxylin and eosin stained brain sections from each mouse were scored blind for degree of vacuolation (ranked 0 to 5) found in nine grey matter regions and three white matter regions (Fraser and Dickinson, 1968). The brain regions analysed were: grey matter areas: (GM1) dorsal medulla, (GM2) cerebellar cortex, (GM3) superior colliculus, (GM4) hypothalamus, (GM5) thalamus, (GM6) hippocampus, (GM7) septum, (GM8) retrosplenial and adjacent motor cortex, (GM9) cingulate and adjacent motor cortex; white matter areas: (WM1) cerebellar white matter, (WM2) mesencephalic tegmentum, (WM3) pyramidal tract. Mean scores were calculated for the RIII mouse groups to produce lesion profiles.
Western blot analysis
Frozen brain samples were homogenized using a micro-pestle in nine volumes of Tris-buffered saline (pH 7.6), containing 0.5% Nonidet™ P40 and 0.5% sodium deoxycholate, to give a 10% w/v suspension. This material was cleared by centrifugation at 420 g for 5 min at 4°C and the supernatant treated with 50 µg/ml proteinase K for 1 h at 37°C. For further details see Head et al., (2004). For samples with low levels of PrPSc the proteinase K treated material (0.1 ml) was centrifuged at 14 000 rpm for 1 h at 4°C, the supernatant discarded, then the pellet resuspended in 0.02 ml loading buffer. The digested product was heat denatured for 10 min at 100°C then loaded onto a 10% Bis/Tris NuPAGE® Novex gel (Invitrogen). After electrophoresis the gel was blotted onto polyvinylidene difluoride membrane. Detection of prion protein used the ECL+ technique (Amersham Biosciences) with primary antibody 6H4 (Prionics) at 1:40 000 and an anti-mouse IgG peroxidase-linked secondary antibody (Amersham Biosciences) at 1:40 000. Images were captured on X-ray film and using a Bio-Rad ChemiDoc XRS+ imaging system. Western blot analysis of proteinase K-treated PrPSc, detected with the 6H4 antibody, typically results in three bands of unglycosylated, monoglycosylated and diglycosylated fragments with the highest molecular weight diglycosylated band running at ∼30 kDa and lowest molecular weight unglycosylated band running at 21 kDa (termed type 1) or 19 kDa (termed type 2) (Head et al., 2004).
Results
Inoculation of brain tissue homogenate from MM blood donor and MV blood recipient
Inoculation of brain tissue homogenate from the variant Creutzfeldt–Jakob disease blood donor resulted in clinical disease and pathology in both HuMM and RIII mouse lines (Table 1, Figs 1 and 2). Incubation times were similar to those previously observed in these mouse lines following inoculation of brain tissue homogenate from other patients with variant Creutzfeldt–Jakob disease (Bruce et al., 2001; Bishop et al., 2006; Ritchie et al., 2009; Diack et al., 2012).
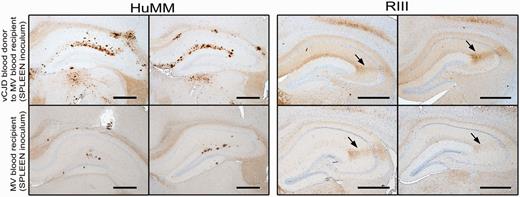
Comparison of immunocytochemistry data from HuMM and RIII mice inoculated with brain and spleen derived inoculum from the MM variant Creutzfeldt–Jakob (vCJD) disease blood donor, and spleen derived inoculum from the MV blood recipient. Scale bar = 200 µm; anti-prion protein antibody 6H4; arrow = CA2 region.
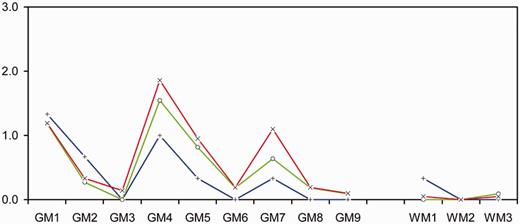
Comparison of lesion profile data from RIII mice. Red line (×) = MM blood donor brain (n = 21); blue line (+) = MM blood donor spleen (n = 3); green line (O) = MV blood recipient spleen (n = 11); brain regions are listed in the ‘Materials and methods’ section.
Primary inoculation into HuMM transgenic and RIII wild-type mouse lines
| Inoculum source | HuMM | RIII | |
|---|---|---|---|
| Clinical positive and incubation period | |||
| MM blood donor | Brain | 2/17 (529 days) | 18/28 (404 days) |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 0/18 | 5/21 (680 days) |
| MV blood recipient | Spleen | 1/18 (611 days) | 12/23 (609 days) |
| Vacuolation-positive | |||
| MM blood donor | Brain | 7/17 | 21/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 3/18 | 3/21 |
| MV blood recipient | Spleen | 1/18 | 11/22 |
| Immunocytochemistry- positive for PrPSc | |||
| MM blood donor | Brain | 16/16 | 25/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 16/17 | 7/20 |
| MV blood recipient | Spleen | 9/19 | 14/23 |
| Inoculum source | HuMM | RIII | |
|---|---|---|---|
| Clinical positive and incubation period | |||
| MM blood donor | Brain | 2/17 (529 days) | 18/28 (404 days) |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 0/18 | 5/21 (680 days) |
| MV blood recipient | Spleen | 1/18 (611 days) | 12/23 (609 days) |
| Vacuolation-positive | |||
| MM blood donor | Brain | 7/17 | 21/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 3/18 | 3/21 |
| MV blood recipient | Spleen | 1/18 | 11/22 |
| Immunocytochemistry- positive for PrPSc | |||
| MM blood donor | Brain | 16/16 | 25/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 16/17 | 7/20 |
| MV blood recipient | Spleen | 9/19 | 14/23 |
Primary inoculation into HuMM transgenic and RIII wild-type mouse lines
| Inoculum source | HuMM | RIII | |
|---|---|---|---|
| Clinical positive and incubation period | |||
| MM blood donor | Brain | 2/17 (529 days) | 18/28 (404 days) |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 0/18 | 5/21 (680 days) |
| MV blood recipient | Spleen | 1/18 (611 days) | 12/23 (609 days) |
| Vacuolation-positive | |||
| MM blood donor | Brain | 7/17 | 21/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 3/18 | 3/21 |
| MV blood recipient | Spleen | 1/18 | 11/22 |
| Immunocytochemistry- positive for PrPSc | |||
| MM blood donor | Brain | 16/16 | 25/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 16/17 | 7/20 |
| MV blood recipient | Spleen | 9/19 | 14/23 |
| Inoculum source | HuMM | RIII | |
|---|---|---|---|
| Clinical positive and incubation period | |||
| MM blood donor | Brain | 2/17 (529 days) | 18/28 (404 days) |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 0/18 | 5/21 (680 days) |
| MV blood recipient | Spleen | 1/18 (611 days) | 12/23 (609 days) |
| Vacuolation-positive | |||
| MM blood donor | Brain | 7/17 | 21/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 3/18 | 3/21 |
| MV blood recipient | Spleen | 1/18 | 11/22 |
| Immunocytochemistry- positive for PrPSc | |||
| MM blood donor | Brain | 16/16 | 25/28 |
| MV blood recipient | Brain | 0/18 | 0/21 |
| MM blood donor | Spleen | 16/17 | 7/20 |
| MV blood recipient | Spleen | 9/19 | 14/23 |
In contrast, there was no clinical or pathological evidence for transmission of disease from inoculum derived from the MV blood recipient brain tissue in either HuMM or RIII mice. Immunohistochemical analysis of the HuMM and RIII mice inoculated with the blood donor brain homogenate showed a variability in amount of PrPSc but similar distribution pattern of PrPSc in the brain, to previously published variant Creutzfeldt–Jakob disease transmission studies (Bishop et al., 2006; Ritchie et al., 2009; Diack et al., 2012).
Western blot analysis of PrPSc extracted from the brains of both HuMM and RIII mice inoculated with the variant Creutzfeldt–Jakob disease blood donor brain material showed a type 2 mobility (of the lower unglycosylated fragment) as expected for variant Creutzfeldt–Jakob disease transmissions to these mice (Figs 3 and 4). PrPSc was not detectable in mice inoculated with the MV blood recipient brain tissue (HuMM data shown in Fig. 3, Lane 4).
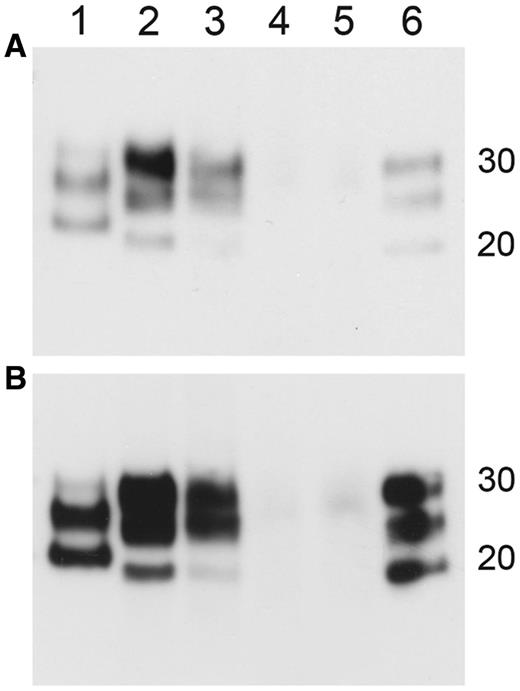
Western blot data from HuMM mice inoculated with brain and spleen derived variant Creutzfeldt–Jakob disease inocula. Duplicate blots (A and B) exposed for different lengths of time to visualize band mobility. Lanes: (1) sporadic Creutzfeldt–Jakob disease type 1; (2) blood donor brain tissue inoculum; (3) blood donor spleen tissue inoculum; (4) blood recipient brain tissue inoculum; (5) blood recipient spleen tissue inoculum; (6) variant Creutzfeldt–Jakob disease type 2B. Molecular weight marker position shown at 20 kDa and 30 kDa. Antibody = 6H4.
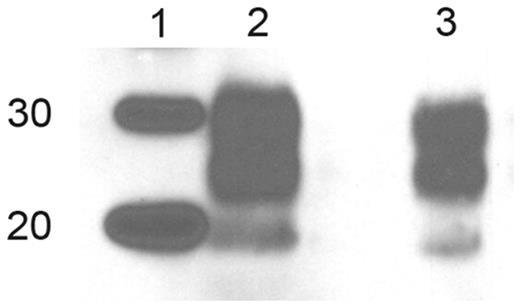
Western blot data from RIII mice inoculated with brain and spleen derived variant Creutzfeldt–Jakob disease inocula. Lanes: (1) molecular weight marker (20 kDa and 30 kDa); (2) blood donor brain tissue inoculum; (3) blood recipient spleen tissue inoculum. Antibody = 6H4.
Thus, there was no evidence of transmissible variant Creutzfeldt–Jakob disease infection in the sample of brain tissue from the MV genotype blood transfusion recipient, while the brain tissue homogenate from the variant Creutzfeldt–Jakob disease blood donor has similar transmission properties to previously reported cases with variant Creutzfeldt–Jakob disease (Bruce et al., 2001; Bishop et al., 2006; Ritchie et al., 2009; Diack et al., 2012). This lack of transmission adds further evidence to the hypothesis that the variant Creutzfeldt–Jakob disease infection had not reached the CNS as initially proposed by the absence of variant Creutzfeldt–Jakob disease pathology in the brain of this patient (Peden et al., 2004).
Inoculation of spleen tissue homogenate from the MM blood donor and MV blood recipient showed similar transmission properties
Clinical signs and/or transmissible spongiform encephalopathy pathology were present in RIII and HuMM mice inoculated with the MM blood donor spleen homogenate and the MV blood recipient spleen homogenate (Table 1, Figs 1 and 2). A comparison of incubation periods between MM blood donor spleen and brain inocula suggested a lower infectivity titre in the spleen, as reported previously (Ritchie et al., 2009).
A lower proportion of RIII mice were scored positive for clinical and pathological signs with the MM blood donor spleen inoculum in comparison to the MV blood recipient spleen, suggesting a higher titre of infectivity in the spleen of the MV blood recipient. However, only 1 of 18 of the HuMM transgenic mice inoculated with the MV blood recipient spleen inoculum was positive for transmissible spongiform encephalopathy vacuolation in the brain whereas 3 of 18 HuMM mice inoculated with the MM blood donor spleen inoculum were positive. One potential explanation for these differences is variation in levels of infectivity resulting from differences in tissue sampling. However, this is an unlikely explanation since the transgenic and wild-type mice gave opposite results. An alternative explanation is that the infectivity within the spleen of the MV blood recipient replicated less efficiently in the HuMM mice.
Lesion profiles for the RIII mice indicated that inoculation of blood donor and recipient spleen tissue resulted in similar pathological targeting in the brain (Fig. 2). This lesion profile is similar to that observed in RIII mice after inoculation with variant Creutzfeldt–Jakob disease infected brain material (Ritchie et al., 2009). Lesion profiles are usually generated from a minimum of five mice and, although data from only three mice inoculated with the MM blood donor spleen tissue were available, the mean scores closely follow the pattern seen for the MV blood recipient spleen inoculum. The three characteristic peaks in vacuolation intensity were for the brain regions: GM1 (dorsal medulla), GM4 (hypothalamus) and GM7 (septum).
Although there was a degree of variation between individual mice, there were no major differences in either the distribution (targeting) or morphology of the PrPSc deposits in the brains of HuMM and RIII mice inoculated with spleen homogenate from the MM blood donor and MV recipient (Fig. 1). RIII mice exhibited more widespread PrPSc deposition than the HuMM, with a granular pattern of deposition, and intense staining in the CA2 region of the hippocampus; the thalamus; the medulla; and the pons. Plaque-like aggregates of PrPSc in the molecular layer of the CA1 region of the hippocampus and more diffuse staining in white matter fibres were seen in HuMM mice. All these deposition patterns have been seen before in previous studies and are characteristic of the transmission properties of variant Creutzfeldt–Jakob disease brain and lymphoid tissues (Fig. 1) (Brown et al., 2003; Bishop et al., 2006; Ritchie et al., 2009).
PrPSc deposited in the brains of the HuMM mice inoculated with the blood donor brain and spleen tissue both gave a type 2 mobility unglycosylated band pattern on western analysis (Fig. 3). In HuMM mice, the amount of PrPSc detected by immunocytochemistry analysis following inoculation of the MV blood recipient spleen is low and extremely focal when compared with that observed with the MM blood donor spleen inoculum (Fig. 1) and as expected proved to be too low for detection by western analysis even when using concentrating methods of preparation (Fig. 3, Lane 5). Samples of brain tissue homogenate from HuMM mice inoculated with the MV blood recipient brain tissue, that were negative by immunocytochemistry, were also confirmed as negative by this concentration method (Fig. 3, Lane 4). Western analysis of the brains of the infected RIII mice demonstrated that both MV blood recipient spleen tissue inoculation and MM blood donor brain inoculation showed a similar type 2 mobility unglycosylated band pattern (Fig. 4). Tissue samples for western analysis were not available from RIII mice infected with the MM blood donor spleen tissue and samples from RIII mice infected with the MV blood recipient brain tissue were negative by the concentration method.
Discussion
This study provides definitive evidence that spleen tissue from an asymptomatic individual contains variant Creutzfeldt–Jakob disease infectivity and that the variant Creutzfeldt–Jakob disease agent retains infectivity following passage through an MV genotype host. The findings are of importance as there has been uncertainty as to whether prion protein immunostaining in peripheral tissues from non-clinical variant Creutzfeldt–Jakob disease necessarily correlated with infectivity (Hilton et al., 2004; Wadsworth et al., 2011). The demonstration of infectivity in such tissues underlines the potential for asymptomatic carriers of variant Creutzfeldt–Jakob disease infection to pose a risk of secondary transmission of infection through blood transfusion or contamination of surgical instruments. The incubation times recorded in this study would suggest moderate but significant levels of infectivity are present in the spleen of the MV genotype recipient, raising the possibility that other peripheral tissues and the blood of this individual are also infected, as indicated by immunohistochemistry in the initial report of this case (Peden et al., 2004). Furthermore the confirmation that there is prion infectivity in an individual with a PRNP codon 129 MV genotype indicates that this genetic subgroup, which accounts for 50% of the UK population, can act as carriers of variant Creutzfeldt–Jakob disease infection.
Our results also provide initial evidence that the variant Creutzfeldt–Jakob disease transmission properties in the MV blood recipient spleen tissue are similar to those of the MM blood donor. This is a critical issue for public health as there is a potential for these infectious agents to change characteristics, including virulence, after serial transmission or following passage in a different genetic background. The relative stability of the agent also makes it more likely that the clinical phenotype of variant Creutzfeldt–Jakob disease infection in an MV background may be similar to that of the well-recognized clinical phenotype in an MM background. While a first passage of an agent between species is not normally sufficient to confirm strain identity, the similarities identified at first passage in this study are striking. We are now undertaking a more extensive strain comparison by our standard serial-passage method using inocula derived from this primary transmission experiment (Ritchie et al., 2009). This will help establish whether the strain characteristics have indeed remained stable following passage through an MV host.
This study has established that variant Creutzfeldt–Jakob disease infectivity can be replicated within a PRNP codon 129 MV genotype host and within a non-CNS tissue, in the absence of variant Creutzfeldt–Jakob disease pathology in the brain. This demonstrates the potential for asymptomatic carriage of variant Creutzfeldt–Jakob disease infection in the UK population, underlining the risk of a silent subclinical epidemic that could result from transfer of infection through blood transfusion or surgery (Garske and Ghani, 2010). It is imperative therefore that continued active surveillance and infection control measures for variant Creutzfeldt–Jakob disease are continued into the future.
Funding
This is an independent report commissioned and funded by the Policy Research Programme in the Department of Health, United Kingdom. The views expressed in the publication are those of the authors and not necessarily those of the Department of Health. Work undertaken at The Roslin Institute was also funded by the Medical Research Council. The tissue bank in the National CJD Research & Surveillance Unit is supported by the Medical Research Council (G1100616).
Acknowledgements
We gratefully acknowledge the assistance of the Biological Resource Facility staff for clinical assessment of the mice and the Pathology Division for sectioning the mouse brains and assessing the levels of transmissible spongiform encephalopathy vacuolation. Mark Head, Laura McCulloch and Allister Smith gave assistance with the analysis of western blots. We also wish to thank Enrico Cancellotti for many discussions throughout this work.
Abbreviations
- HuMM
transgenic mouse line expressing human prion protein with MM genotype at codon 129
- PrPSc
disease associated (‘scrapie’) form of the prion protein
References
Author notes
*These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}