




Abstract
Machado-Joseph disease or spinocerebellar ataxia type 3, the most common dominantly-inherited spinocerebellar ataxia, results from translation of the polyglutamine-expanded and aggregation prone ataxin 3 protein. Clinical manifestations include cerebellar ataxia and pyramidal signs and there is no therapy to delay disease progression. Beclin 1, an autophagy-related protein and essential gene for cell survival, is decreased in several neurodegenerative disorders. This study aimed at evaluating if lentiviral-mediated beclin 1 overexpression would rescue motor and neuropathological impairments when administered to pre- and post-symptomatic lentiviral-based and transgenic mouse models of Machado-Joseph disease. Beclin 1-mediated significant improvements in motor coordination, balance and gait with beclin 1-treated mice equilibrating longer periods in the Rotarod and presenting longer and narrower footprints. Furthermore, in agreement with the improvements observed in motor function beclin 1 overexpression prevented neuronal dysfunction and neurodegeneration, decreasing formation of polyglutamine-expanded aggregates, preserving Purkinje cell arborization and immunoreactivity for neuronal markers. These data show that overexpression of beclin 1 in the mouse cerebellum is able to rescue and hinder the progression of motor deficits when administered to pre- and post-symptomatic stages of the disease.
Introduction
The autosomal dominant spinocerebellar ataxias (SCAs) are a complex group of neurodegenerative disorders that comprise 28 different genetic loci and are characterized by progressive cerebellar ataxia of gait and limbs along with other variable symptoms. Among the SCAs, six are known to be polyglutamine diseases (SCA1–3, SCA6–7 and SCA17) (Duenas et al., 2006; Carlson et al., 2009).
Machado–Joseph disease, also known as spinocerebellar ataxia type 3 (SCA3) is the most common autosomal dominant SCA and one of the most common polyglutamine diseases worldwide (Schols et al., 2004). The disease is caused by a genetic mutation involving over-repetition of the trinucleotide CAG in the MJD1/ATXN3 gene, which translates into an expanded polyglutamine tract within the ataxin 3 protein (Kawaguchi et al., 1994). This protein adopts an abnormal folding and has an increased propensity to aggregate (Paulson et al., 1997). The expanded polyglutamine tract is shared among the polyglutamine diseases, which accounts for the clinical and neuropathological similarities observed in these diseases (Bauer and Nukina, 2009). Machado–Joseph disease is a multisystem neurodegenerative disorder characterized by a wide range of clinical manifestations that include: limb and gait cerebellar ataxia; pyramidal syndrome; peripheral neuropathy and extra-pyramidal signs including dystonia, rigidity and/or bradykynesia, among others (Rosenberg, 1992; Sudarsky and Coutinho, 1995). The clinical manifestations appear late in life and there is growing evidence that to this may contribute a progressive failure of protein degradation systems (Cuervo, 2004).
Beclin 1 was among the first mammalian autophagy effectors to be identified (Liang et al.1998). It is a key protein of the autophagy clearance pathway, whose activity is inhibited by binding of Bcl-2 homologs to its BH3 domain. In mammalian cells, beclin 1 interacts with the class III PI3K to activate the nucleation step of autophagy, also playing a role in the fusion of autophagosomes to lysosomes (Itakura et al., 2008; Sun et al., 2008; Matsunaga et al., 2009; Zhong et al., 2009). Beclin 1 appears to play a general role in cell survival. In mice, homozygous deletions (Becn1−/−) lead to death at an early embryonic stage (Qu et al., 2003; Yue et al., 2003) while heterozygous deletions (Becn1+/−) lead to an increased frequency of spontaneous lymphomas and carcinomas (Qu et al., 2003; Yue et al., 2003) as well as alterations in neuronal autophagy and neuronal function (Pickford et al., 2008). On the contrary, overexpression of beclin 1 in the brain of adult mice has been shown to protect neurons from apoptosis induced by Sindbis virus infection (Liang et al., 1998) as well as from accumulation of misfolded proteins (Pickford et al., 2008; Jaeger et al., 2010). In previous work, we and others have reported that levels of beclin 1 were decreased upon ageing, Alzheimer's disease, Huntington's disease and Machado–Joseph disease (Shibata et al., 2006; Pickford et al., 2008; Nascimento-Ferreira et al., 2011), wheras beclin 1 overexpression increased the autophagic clearance of misfolded amyloid-β (Pickford et al., 2008; Jaeger et al., 2010), alpha-synuclein (Spencer et al., 2009) and ataxin 3 protein (Nascimento-Ferreira et al., 2011) and at the same time promoted neuroprotection. Nevertheless, it has not yet been proved if these neuroprotective effects of beclin 1 are translated into motor improvements, which would be a major asset to these highly motor-incapacitating diseases.
From a translational perspective it is important to investigate if beclin 1 overexpression per se may improve neuropathology and motor behaviour in mice already severely affected by the movement disorder, an outcome that is relevant for polyglutamine diseases and SCAs in general. In fact, several studies have recently shown that some drugs used in cancer therapy, such as imatinib (Can et al., 2011), lapatinib (Huang et al., 2011) and sunitinib (Zhao et al., 2010), can upregulate beclin 1, opening a new therapeutic window for these drugs and hopefully, for several neurodegenerative diseases.
Therefore, in this work we aimed at evaluating whether beclin 1 would be able to improve motor and neuropathological deficits when administered to two different mouse models: a lentiviral-based model of Machado–Joseph disease presymptomatic at the time of initiation of beclin 1 expression, and a transgenic mouse model expressing a truncated fragment of mutant ataxin 3 (Torashima et al., 2008), symptomatic and with cerebellar atrophy since the beginning of the study.
Beclin 1 was able to hamper the development of motor deficits and neuropathology when administered to the lentiviral mouse model, improving motor coordination, balance, gait and expression of neuronal markers to a level similar to wild-type mice. In addition, beclin 1 was also able to mediate improvements in the motor function as well as a decrease in the cerebellar atrophy when administered after onset of the pathology to transgenic mice resembling a late and severe phenotype of cerebellar ataxia. This study shows that beclin 1 overexpression hinders the progression of motor and neuropathological abnormalities when administered to pre- and postsymptomatic stages of Machado-Joseph disease, an observation not only relevant to polyglutamine diseases and cerebellar ataxias but also to other neurodegenerative diseases.
Materials and methods
Lentiviral vectors
Viral vectors encoding for GFP (Regulier et al., 2002), human beclin-1, EGFP-LC3 (Nascimento-Ferreira et al., 2011) and human full-length mutant ataxin 3 with 72 glutamines (Atx3Q72) (Alves et al., 2008), were produced in human embryonic kidney (HEK) 293T cells using a four-plasmid system described previously (de Almeida et al., 2001, 2002). Viral stocks were stored at −80°C until use.
Primary cerebellar cell culture
The primary cerebellar cell culture was isolated from transgenic Q69 (confirmed by genotyping) within 6–7 days after birth. Cells were cultured with a density of 500 000 cells/ml in Neurobasal® medium (Gibco, Life Technologies), supplemented with l-glutamine (Sigma) (0.5 mM), gentamicin (Gibco, Life Technologies) (50 µg/ml), B-27 supplement (Gibco, Life Technologies), and potassium chloride (Sigma) (2 M). Cells were plated and maintained in multiwells previously coated with poly-l-lysine (Sigma), at 37°C in 5% CO2/air atmosphere.
Cerebellar cell culture infection
For the infection of cerebellar cells culture, the medium was replaced with 400 µl of new complete medium and 1.6 µl of hexadimethrine bromide (Sigma-Aldrich) (8 µg/ml) and the corresponding lentivirus added (10 ng/100 000 cells) according to the condition (beclin 1 or GFP as control). The multiwells were then incubated at 37°C in 5% CO2/air atmosphere, and after 6 h of infection, 600 µl of new fresh medium (complete) was added to the wells. At 2 weeks post-infection cells were lysed for western blotting analysis.
Neuroblastoma cell culture
Mouse neuroblastoma cell line (Neuro-2A cells) obtained from the American Type Culture Collection cell biology bank (CCL-131) were incubated in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin (Gibco) (complete medium) at 37°C in 5% air atmosphere.
Neuroblastoma cell cultures infection and drug treatment
Cells were plated and infected with lentiviral particles expressing enhanced green fluorescent protein-light chain 3 (EGFP) with or without beclin-1 lentiviral vector at the ratio of 10 ng of p24 antigen/105 cells. At 2 weeks post-infection, cells were treated with rapamycin (0.2 µM) for 6 h or control (dimethyl sulphoxide) in the presence or absence of chloroquine (100 µM) for 6 h. The cells were then fixed for microscopy and lysed for western blot.
Neuroblastoma cell cultures imaging and quantification
Cells expressing EGFP-LC3 alone, co-expressing beclin 1 or treated with rapamycin were analysed by confocal microscopy (2 weeks after infection). Chloroquine, a drug known to raise the lysosomal pH, leading to inhibition of both fusion of autophagosomes with lysosomes and lysosomal protein degradation (Shintani and Klionsky, 2004) was used to allow autophagosome accumulation and quantification. LC3 puncta and size were quantified using the ‘analyse particles’ function of the ImageJ software after thresholding of images with size settings from 0.2–10 pixel2 and a circularity of 0–1. At least 15 cells were analysed for each condition. Values are expressed as mean ± SEM of threee independent experiments.
Protein extraction and western blotting
Brains and cells were lysed in RIPA-buffer solution (50 mM Tris HCl pH 8, 150 nM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitors (Roche diagnostics) followed by a 4 s ultra-sound chase (1 pulse/s). Protein concentration was determined with the Bradford protein assay (Bio-Rad). Sixty micrograms of protein extract were resolved in SDS-polyacrylamide gels (12%). The proteins were transferred onto PVDF membrane (GE Healthcare) according to standard protocols. The membranes were blocked by incubation in 5% non-fat milk powder in 0.1% Tween 20 in Tris buffered saline (TBS-T) for 1 h at room temperature, and were then incubated overnight at 4°C with mouse monoclonal anti-beclin 1 (BD Biosciences; 1:1000), mouse monoclonal anti-β-actin (Sigma; 1:5000), rabbit anti-LC3 (Cell Signaling; 1:1000), mouse anti-α-tubulin (Sigma; 1:5000) antibodies. Blots were washed three times in TBS-T, for 15 min each, and incubated with the secondary antibody goat anti-mouse or anti-rabbit (1:10 000; Vector Laboratories) for 2 h at room temperature. Blots were washed three times in TBS-T, for 15 min each and the bands were visualized with Enhanced Chemifluorescent substrate (ECF) (GE Healthcare) and chemifluorescence imaging (Versadoc Imaging System Model 3000, Bio-Rad). Semi-quantitative analysis was carried out based on the optical density of scanned membranes (Quantity One® 1-D image analysis software version 4.4; Bio-Rad). The specific optical density was then normalized with respect to the amount of β-actin or alpha-tubulin loaded in the corresponding lane of the same gel.
Flow cytometry analysis
Wild-type mice with 21 days of age were stereotaxically-injected in the cerebellar cortex with lentiviral vectors encoding EGFP-LC3 (n = 4) or EGFP-LC3 plus beclin 1 (1:1; n = 3). Two weeks post-injection mice were sacrificed and transcardiacally perfused with a phosphate buffer solution. Afterwards, the brain was removed, the cerebellar cortex isolated and homogenized using a glass dounce homogenizer. The homogenate solution was then filtered through a 70/40 µm cell strainer into a 50 ml Falcon tube to obtain a single-cell suspension. Centrifugation at 1200 rpm for 5 min at room temperature was performed twice to pellet and wash the cells in PBS. The final pellet was resuspended in 1 ml of PBS to be sequentially analysed by flow cytometry. Samples were kept on ice until use, and analysis of 100 000 cells per animal was performed on a fluorescence-activated cell-detector flow cytometer (Becton Dickinson). Data of viable cell counts was plotted as GFP fluorescence intensity (FL1 channel, 530 ± 30 nm).
In vivo experiments
Transgenic mouse model
Polyglutamine disease transgenic mouse models expressing the N-terminal-truncated ataxin 3 with 69 glutamine repeats and an N-terminal haemagglutinin epitope driven by Purkinje-cell-specific L7 promoter (Tg-Q69) were previously described (Torashima et al., 2008; Oue et al., 2009). A colony of these transgenic mice was established in the Centre for Neuroscience and Cell Biology of the University of Coimbra and the line was maintained by backcrossing heterozygous males with C57BL/6 females. Genotyping was performed by PCR. The present study used eight heterozygous females and eight heterozygous males divided in two groups of animals injected with lentiviral vectors encoding GFP (Tg-Q69+ control; n = 8) and beclin 1 (Tg-Q69+ beclin 1; n = 8), respectively. The wild-type littermates, 16 males and 14 females were used for control (wild-type, n = 8) and for the lentiviral-based Machado–Joseph’s disease model. The behavioural testing began at 21–25 days of age (P21–25). At this age, transgenic mice were already phenotypically distinguishable from their wild-type littermates by home-cage behaviour.
Lentiviral Machado–Joseph disease mouse model
Experiments with the lentiviral Machado–Joseph disease mouse model, previously characterized (Nobrega et al., 2012), involved injection of a mix of lentiviral vectors (1:1) encoding, in the control group, the full-length mutant ataxin 3 with 72 glutamine repeats and GFP (LV-Atx3Q72+ control; n = 8) and in the other group, the full-length mutant ataxin 3 with 72 glutamine repeats and beclin 1 (LV-Atx3Q72+ beclin 1; n = 8). The behavioural testing began at 21–25 days of age (P21–25). All animals were housed in a temperature-controlled room and maintained on a 12 h light/dark cycle. Food and water were available ad libitum. The experiments were carried out in accordance with the European Community Council directive (86/609/EEC) for the care and use of laboratory animals.
Stereotaxic surgery
Concentrated viral stocks were thawed on ice and resuspended by vortexing. Mice were anaesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) with xylazine (10 mg/kg). Particle content of lentiviral vectors was matched to 200 000 ng of p24/ml. The animals received a single injection of 6 µl of lentiviral vectors (6 µl of beclin 1/GFP in Tg-Q69 mice; 3 + 3 µl of mutant Atx3+ beclin 1/GFP in LV-Atx3Q72 mice) at a rate of 0.25 µl/min by means of an automatic injector (Stoelting Co.), at the following coordinates: −1.6 mm rostral to lambda, 0.0 mm midline, and −1.0 mm ventral to the skull surface, with the mouth bar set at −3.3. After the injection, the syringe needle was left in place for an additional 5 min to minimize backflow.
Behavioural assessment
Mice were trained on a battery of motor tests starting at 21–25 days of age (P21–25), before surgery (T0) and performed every 2 weeks until 10 weeks. All tests were performed in a dark room after 30 min of acclimatization.
Modified beam test
Motor coordination and balance were assessed by measuring the ability of mice to hold or walk for 15 s in long wood beams (1 m) of 0.5 and 0.8 mm round diameter or 1 mm of square cross section. The beams were placed horizontally, 50 cm above a padded surface, with both ends mounted on a support. The test started with the most difficult beam (0.5 mm round) and ended with the easiest beam (1 mm square). The score given to each animal corresponded to the beam in which they could perform the test.
Rotarod
The Rotarod apparatus (Letica Scientific Instruments) was used to measure fore and hind limb motor coordination and balance. Each mouse was placed on the Rotarod at a constant speed (5 rpm) for a maximum of 5 min, and at an accelerated speed (4 to 40 rpm in 5 min) for a maximum of 5 min. The latency to fall was recorded. Mice were allowed to perform four trials for each test and time point, with 15 min rest between trials. For analysis, the mean latency to fall off the Rotarod of three to four trials was used.
Footprint test
The footprint test was used to compare the gait of beclin 1 and control-treated lentiviral and transgenic Machado–Joseph disease mice. To obtain the footprints, the mice hind- and forefeet were coated with black and green non-toxic paints, respectively. The animals were then allowed to walk along a 100-cm long, 10-cm wide runaway (with 15-cm high walls). A fresh sheet of white paper was placed on the floor of the runaway for each mouse run. The footprint patterns were analysed for four step parameters (all measured in centimetres). Stride length was measured as the average distance of forward movement between each stride. Hind-base width and front-base width were measured as the average distance between left and right hind footprints, respectively. These values were determined by measuring the perpendicular distance of a given step to a line connecting its opposite preceding and proceeding steps. Distance from left or right front footprint/hind footprint overlap was used to measure uniformity of step alternation. When the centre of the hind footprint fell on top of the centre of the preceding front footprint, a value of zero was recorded. When the footprints did not overlap, the distance between the centre of the footprints was recorded. A sequence of six consecutive steps was chosen for evaluation, excluding footprints made at the beginning and end of the run where the animal was initiating and finishing movement, respectively. The same operator made all footprint measurements. The mean value of each set of five to six values was used for analysis.
Histological processing
Tissue preparation
Animals were killed by sodium pentobarbital overdose, transcardially perfused with a 4% paraformaldehyde fixative solution (PFA 4%, Fluka) followed by brain removal. After a cryoprotective incubation in 25% sucrose, 0.1 M PBS for 48 h, brains were frozen in dry ice (−80°C) and 20 µm coronal sections were cut at a cryostat-microtome (Leica CM3050S, Leica Microsystems). Slices throughout the entire cerebellum were collected in SuperFrost® plus microscope slides (Thermo Fisher Scientific) and stored at −20°C before immunohistochemical processing.
Immunohistochemical procedure
The immunohistochemical procedure was initiated with a 30 min dehydration at 37°C followed by a 30 min hydration in 0.1 PBS and 1 h blocking in a 0.3% Triton™ in 0.1 PBS with 10% normal goat serum both at room temperature. The following primary antibodies diluted in blocking solution with 0.1% Triton™ were used: mouse monoclonal anti-ataxin 3 (1H9, Chemicon; 1:5000; overnight, 4°C), mouse monoclonal anti-haemagglutinin (InvivoGen; 1:1000; overnight, 4°C), mouse monoclonal anti-beclin 1 (BD Biosciences; 1:1000; overnight, 4°C), rabbit polyclonal anti-DARPP-32 (Chemicon; 1:5000, overnight, 4°C) and rabbit polyclonal anti-Calbindin D-28K (Chemicon; 1:1000, overnight, 4°C). Sections were then incubated in secondary antibody, goat-anti rabbit and/or mouse conjugated to Alexa Fluor® 488 or 594 (Invitrogen) for 2 h at room temperature and then mounted in FluorSave™ (Calbiochem) with 4’,6’-diamidino-2-phenylindole (DAPI). Fluorescence images were acquired with a Zeiss Axiovert 200 imaging microscope or LSM Zeiss microscope for double staining experiments.
In vivo imaging quantification
Quantification of ataxin 3-positive inclusions was performed in the lentiviral model after performing 1H9 immunohistochemistry and imaging. Six mice/group (n = 6) were used and four coronal sections spread over the anterior–posterior extent of the cerebellum (inter-section distance: 200 - 300 µm) were scanned using a ×20 objective on a Zeiss Axiovert 200 imaging microscope as well as an image acquisition and analysis software (ImageJ, NIH). For each coronal section and animal eight fields were acquired. The average number of inclusions per section of the cerebellum was calculated for each animal. Quantification of haemagglutinin-tagged ataxin 3-positive inclusions was performed in the transgenic model after performing haemagglutinin immunohistochemistry and imaging. Five mice/group (n = 5) were used and four coronal sections spread over the anterior–posterior extent of the cerebellum (inter-section distance: 200–300 µm) were scanned using a ×40 objective on a Zeiss Axiovert 200 imaging microscope as well as an image acquisition and analysis software (ImageJ, NIH). For each coronal section and animal eight fields were acquired. The average number of inclusions per 100 cells quantified by DAPI staining was calculated for each animal. The quantification of DARPP-32 (now known as PPP1R1B) neuronal expression was performed by scanning four coronal sections spread over the anterior–posterior extent of the cerebellum expressing the DARPP-32 protein using a ×40 objective on a Zeiss Axiovert 200 imaging microscope. Six mice per group were used for the lentiviral model (n = 6) and three mice per group (n = 3) were used for the transgenic model. For each coronal section and animal, eight fields were acquired using the same exposure. Optical densitometry analysis was performed using ImageJ analysis software. Cerebellar regions expressing DARPP-32 revealed to have an increased optical density value when compared to regions not expressing the DARPP-32 protein. Values are represented as the mean value of DARPP-32 optical density per section (in arbitrary units). The quantification of calbindin neuronal expression was performed by scanning four coronal sections spread over the anterior–posterior extent of the cerebellum expressing the calbindin protein using a ×20 objective on a Zeiss Axiovert 200 imaging microscope. Four mice/group were used for both models (n = 4). For each coronal section and animal, eight fields were acquired using the same exposure. Optical densitometry analysis was performed using ImageJ analysis software. Cerebellar regions expressing calbindin were revealed to have an increased optical density value when compared to regions not expressing this protein. Values are represented as the mean value of calbindin optical density per section (in arbitrary units).The quantification of the size of the molecular layer was performed by measuring the width of the molecular layer starting at the Purkinje cells. For that purpose three coronal sections per animal stained with calbindin markers were scanned using a ×20 objective on a Zeiss Axiovert 200 imaging microscope. Measurements were performed in triplicate and results converted in micrometres by means of analysis software (ImageJ, NIH).
Cresyl violet histological staining
Coronal and sagittal brain sections were stained with cresyl violet dye for 2 min, differentiated in acetate buffer pH 3.8 (2.72% sodium acetate and 1.2% acetic acid; 1:4 v/v), dehydrated by passing twice through ethanol and toluene solutions, and mounted with Eukitt® (O. Kindler GmbH & CO).
Quantification of the number of Purkinje cells was performed by scanning four coronal sections spread over the anterior–posterior extent of the cerebellum, using a ×20 objective on a Zeiss Axiovert 200 imaging microscope and an image analysis software (ImageJ, NIH). For each coronal section and mice (n = 4/group) six fields were acquired and Purkinje cells counted in a defined region. Values are represented as the mean number of Purkinje cells per 100 µm ± SEM.
Statistical analysis
Differences among groups over time were evaluated by two-way ANOVA followed by Bonferroni’s post hoc test; differences between three groups at the same time point by one-way ANOVA followed by Bonferroni’s post hoc test; and comparisons between two groups by unpaired Student’s t-test, always using the GraphPad Prism software.
Results
Beclin 1 activates autophagy
We previously showed that beclin 1 stimulates autophagic flux and clearance of mutant ataxin 3 when administered to in vitro and in vivo Machado–Joseph disease models (Nascimento-Ferreira et al., 2011). Here we investigated beclin 1 ability to increase autophagy as compared with rapamycin, a drug commonly used as an autophagy inducer in a non-Machado–Joseph disease model context. For that we infected Neuro-2a cells with a vector encoding EGFP-LC3 with or without beclin 1 vector, treated later with rapamycin or control (dimethyl sulphoxide) in the presence or absence of chloroquine (Fig. 1A–I). Beclin 1 significantly increased EGFP-LC3 puncta formation, an effect that is apparent upon addition of the lysosomotropic agent chloroquine (Fig. 1F and G; n = 3, P < 0.001). Interestingly, beclin 1 mediated a robust increase in LC3 puncta, which was superior to that observed with rapamycin (Fig. 1E and G; n = 3, P < 0.01). To confirm beclin 1-mediated effect as an autophagy enhancer, two further sets of experiments were performed in vitro and in vivo: we measured LC3-II levels in Neuro-2A by western blot (Fig. 1H and I) and EGFP-LC3 levels in lentiviral-injected mice by flow cytometry (Fig. 1J and K; n = 3–4, P < 0.01). In both experiments beclin 1 increased autophagic flux, as highlighted by the increased accumulation of autophagosomes upon chloroquine addition (Fig. 1 H and I) and increased autophagosomal degradation of EGFP-LC3 visualized by flow cytometry (Fig. 1K, n = 3–4, P < 0.01), a technique previously characterized for this purpose (Nascimento-Ferreira et al., 2011).
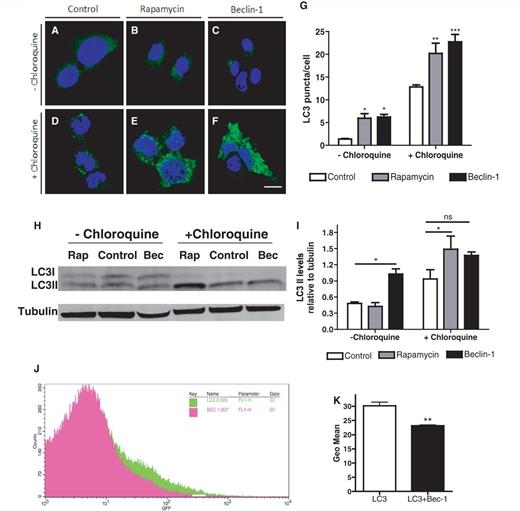
Beclin 1 activates autophagic flux. (A–G) Confocal imaging and quantification. Neuroblastoma cells (Neuro-2A) expressing EGFP-LC3 alone (A and D), co-expressing beclin 1 (C and F) or treated with rapamycin (B and E) were analysed by confocal microscopy in the absence (A–C) or presence of chloroquine (D–F). Quantification of EGFP-LC3-positive puncta (G) confirmed that both rapamycin and beclin 1 increased the number of autophagosomes, an effect particularly apparent in the presence of chloroquine. Values are expressed as mean ± SEM (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). (H–I) Western blot analysis. Neuro-2A expressing beclin 1 (Bec), control (Control) or rapamycin (Rap) in the absence (left lanes) or presence of chloroquine (right lanes) were analysed for endogenous LC3-I and LC3-II levels by western blot (H). Tubulin was used as loading control. Quantification was performed by optical densitometry analysis (I). Each LC3-II levels were normalized according to the tubulin band. Results are expressed as ratio LC3-II/ tubulin. Values are expressed as mean ± SEM (n = 3; *P < 0.05). (J and K) Flow cytometry. EGFP-LC3-injected mice expressing this vector alone (n = 4) or co-expressing beclin 1 (n = 3) were analysed by flow cytometry 2 weeks after injection. Results are expressed as the geometric mean of the fluorescent signal. Values are expressed as mean ± SEM (n = 3–4; **P < 0.01). ns = not significant.
Beclin 1 improves balance and motor coordination deficits in lentiviral–based and transgenic mouse models of polyglutamine disease
Subsequently, we evaluated if beclin 1 was able to (i) hamper the development of motor symptoms when administered to a presymptomatic lentiviral-based Machado–Joseph disease mouse model (Fig. 2A, C, E, G, I and K); and (ii) rescue the motor impairments in a post-symptomatic transgenic mouse model of Machado-Joseph disease and polyglutamine ataxias (Fig. 2B, D, F, H, J and L). In the lentiviral-based Machado–Joseph disease mouse model (LV-Atx3Q72; Fig. 2A, C, E, G, I and K), lentiviral vectors encoding beclin 1 or Green Fluorescent Protein (GFP, control) were co-injected with vectors encoding the full-length mutant ataxin 3 with 72 glutamines (Atx3Q72) in the cerebellum of wild-type mice at 21 days of age (P21). On the other hand, in the transgenic mouse model (Tg-Q69; Fig. 2B, D, F, H, J and L), lentiviral vectors encoding beclin 1 or GFP (control) were administrated to mice already expressing a truncated fragment of mutant ataxin 3 with 69 glutamines (Q69) (Torashima et al., 2008). Importantly, since the beginning of the study (P21) these mice already presented an important cerebellar atrophy (Fig. 2D and F), in particular of the molecular layer of the cerebellum (Fig. 2F), compared with age-matched wild-type mice used to generate the LV-Atx3Q72 (Fig. 2C and E). A single injection of beclin 1 or GFP led to an extensive transduction of the cerebellar cortex (Fig. 2G and H). A significant increase in beclin 1 levels was also observed in beclin 1-injected LVAtx3Q72 mice (Fig. 2I–K; n = 3; 1.24 ± 0.084 versus 0.86 ± 0.11 in control, P = 0.0477) and P7 cerebellar cultures of transgenic mice (Fig. 2J-L; n = 3; 1.67 ± 0.045 versus 0.88 ± 0.10 in control, P = 0.0021).
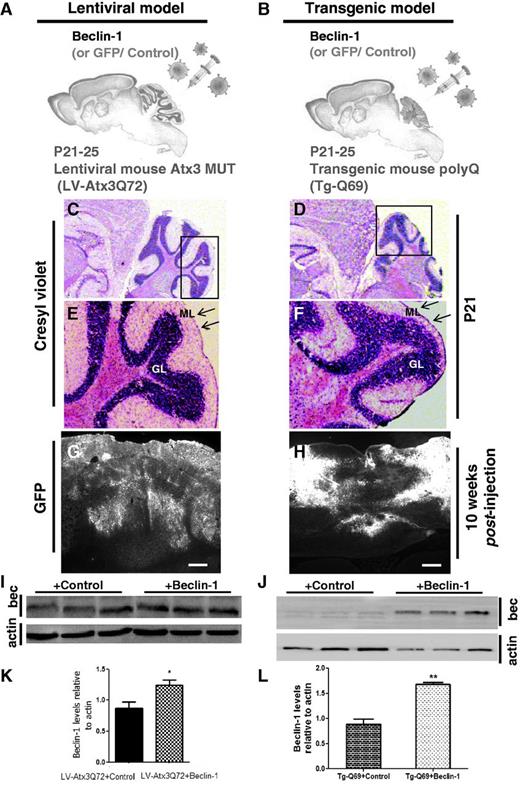
Experimental design and expression of transgenes. (A and B) Experimental design. Wild-type mice between 21–25 days of age (P21–25) were co-injected in the cerebellar vermis with lentiviral vectors (LV) encoding full-length mutant ataxin 3 with 72 glutamine repeats (Atx3Q72) along with beclin 1 or GFP (control) (A). Transgenic mice expressing a fragment of mutant ataxin 3 truncated upstream the 69 glutamine repeats (Tg-Q69), 21–25 days old (P21–25) were injected in the cerebellar vermis with lentiviral vectors encoding beclin 1 or GFP (control) (B). For both models, motor behavioural testing started 1 day before lentiviral-injection (T0) up to 10 weeks post-injection. At that time point, mice were sacrificed and processed for histological analysis. (C–F) Cresyl violet staining. Cresyl violet staining of the lentiviral (C and E) and transgenic model (D and F) at the moment of injection (P21). E and F are high-magnifications of C and D, respectively. Note the cerebellar atrophy present in the transgenic mice at the level of the molecular layer (F, arrows) as compared to the lentiviral mouse model (E, arrows) already at 21 days of age. (G and H) In vivo expression of the transgene. Direct fluorescence of GFP expression at 10 weeks post-injection after a single lentiviral injection in the cerebellar vermis of the lentiviral (G) and transgenic mouse model (H). Scale bar: 200 µm. (I and K) Western blot analysis of mice injected with Atx3Q72 plus beclin 1 or control, showing beclin 1 expression (bec) in the cerebellar lysates 2 weeks post-infection (I). Quantification by optical densitometry revealed a significant increase of beclin 1 levels (K) Actin was used as loading control and results are expressed as ratio beclin 1/actin. Values are expressed as mean ± SEM (n = 3; *P < 0.05). (J and L) Western blot analysis of cerebellar cortex primary cultures derived from Tg-Q69 mice at 7 days of age (P7). Cultures were infected with lenviral vectors enconding GFP (control) or beclin 1 transgene and analysed 2 weeks post-infection (J). Quantification by optical densitometry revealed a significant increase of beclin 1 levels (L). Actin was used as loading control and results are expressed as ratio beclin 1/actin. Values are expressed as mean ± SEM (n = 3; **P < 0.01). ML = molecular layer; GL = granular layer.
Behavioural testing was initiated 1 day before injection of the lentiviral vectors (Fig. 3; T0) and was performed until 10 weeks post-injection (Fig. 3; T10). In the accelerated Rotarod (acceleration from 4 to 40 rotations per min in 5 min), beclin 1-treated LV-Atx3Q72 mice (LV-Atx3Q72+ beclin 1) were rescued from the impairment in balance and motor coordination of GFP-treated mice (LV-Atx3Q72+ control), observed at 10 weeks post-injection (Fig. 3A; n = 7; 120.93 ± 8.07 versus 81.08 ± 4.02 in control, *P < 0.05). Importantly at this time-point, the Rotarod performance of beclin 1-treated LV-Atx3Q72 was indistinguishable from non-injected wild-type mice (Fig. 3A; n = 7–8; 120.93 ± 8.07 versus 120.25 ± 12.60 in wild-type), suggesting a total rescue regarding this test. The Tg-Q69 mouse model showed dramatic motor impairments since the beginning of the test when compared with wild-type mice (Fig. 3B; T0: 17.88 ± 2.32 versus 120.05 ± 6.60 in wild-type, P < 0.001), nevertheless beclin 1 was able to improve the performance of the mice along time, reaching statistical significance at 10 weeks post-injection (Fig. 3B; n = 8; 40.28 ± 9.35 versus 13.95 ± 3.07 in control, P < 0.05). Nevertheless, at that time point the performance of the beclin 1-treated transgenic mice continued to be substantially lower as compared with wild-type. Therefore, to further assess the balance and motor coordination of transgenic mice, the beam test was used. Beclin 1 was able to preserve along time the ability of Tg-Q69 mice to walk or grip the most difficult beams (0.5 and 0.8 cm round) without falling; when compared to control-treated, a significant improvement was observed from 2 weeks post-injection (Fig. 3D; n = 8; T2: 0.54 ± 0.038 versus 0.71 ± 0.067 in control, P < 0.05; T10: 0.54 ± 0.038 versus 0.73 ± 0.049 in control, P < 0.01). In this test, beclin 1-treated transgenic mice showed no statistical difference from non-injected wild-type mice. No differences were observed in the LV-Atx3Q72 model and wild-type (Fig. 3C) as this model had a mild phenotype compared with the transgenic model.
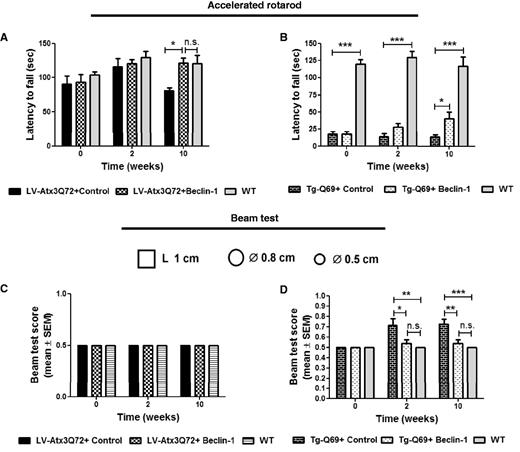
Beclin–1 improves balance and motor coordination deficits in lentiviral based and transgenic mouse models of polyglutamine disease. (A and B) Accelerated Rotarod test. Mice were forced to walk on accelerated rotarod apparatus (4–40 rpm in 5 min). Latency to fall was measured for each time point for the LV-Atx3Q72 model (A) and Tg-Q69 model (B). Beclin 1 administration significantly improved the motor performance of LV-Atx3Q72 mice (A) when compared to GFP/control at 10 weeks post-injection. Note that at this time point beclin 1-treated mice were not statistically different from non-injected wild-type (WT) mice. Overexpression of beclin 1 in Tg-Q69 (B) led to improvement of motor coordination and balance along time, specially at 10 weeks post-injection. Note that the motor performance of these mice was significantly different from non-injected wild-type from the beginning of the study (T0). Results are expressed as mean ± SEM (n = 8; *P < 0.05; ***P < 0.001). (C and D) Modified beam test. The ability to hold for 15 s in a cylindrical 0.5 and 0.8 cm as well as 1 cm square beam was accessed in the lentiviral (C) and transgenic model (D). The score number reveals the beam in which mice were able to perform the test. No differences were observed between the lentiviral model and wild-type mice regarding this test (C). On the other hand control-treated Tg-Q69 mice showed lower performances when performing this test when compared to beclin 1-injected or non-injected wild-type mice already at 2 weeks post-injection (D). Note that beclin 1- treated mice are not statistically different from non-injected wild-type. Values are expressed as mean ± SEM (n = 8; *P < 0.05; **P < 0.01; ***P < 0.001).
Altogether, these results show that beclin 1 overexpression when initiated at the same time as induction of pathology (LV-Atx3Q72) is able to completely rescue the motor coordination and balance skills, while a partial rescue is achieved upon intervention at a late stage of the disease (Tg-Q69).
Beclin 1 hampers the development and progression of ataxic phenotype
An important characteristic of Machado–Joseph disease, as well as of other SCAs and/or polyglutamine diseases is the development of gait and limb ataxia (Carter et al., 1999; Goti et al., 2004; Chen et al., 2008). For that reason, we next analysed footprint patterns to evaluate whether beclin 1 was able to delay the appearance of the ataxic phenotype in the LV-Atx3Q72 (Fig. 4A, C, E and G) and Tg-Q69 model (Fig. 4B, D, F and H). Strikingly, at the 10 week time-point beclin 1-treated LV-Atx3Q72 mice were indistinguishable from wild-type mice regarding stride length (Fig. 4A; n = 8; T10: 7.36 ± 0.18 versus 6.30 ± 0.13 in control, P < 0.001), front base width (Fig. 4C; n = 8; T10: 1.30 ± 0.07 versus 1.54 ± 0.03, P < 0.01), hind base width (Fig. 4E; n = 8; T10: P > 0.05) and distance between front and hind limb overlap in consecutive footsteps (Fig. 4G; n = 8; T10: 0.68 ± 0.07 versus 1.00 ± 0.09, P < 0.05). In the opposite, control GFP-treated LV-Atx3Q72 mice exhibited significant impairments in all parameters, displaying shorter stride length, wider front base width and increased distance in the footprint overlap.
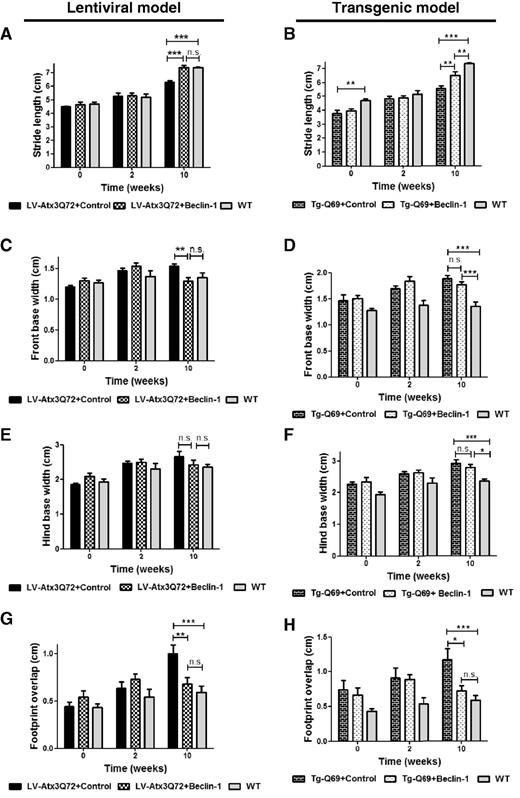
Beclin–1 improves the ataxic phenotype when administered before and after onset of the disease. (A–H) Footprint pattern quantitative analysis. (A and B) Stride length relative to hind paw of LV-Atx3Q72 (A) and of Tg-Q69 model (B). Analyses show that beclin 1-treated mice have a longer stride length. Values are expressed as mean ± SEM (n = 8; **P < 0.01; ***P < 0.001; n.s. = no significant difference). (C and D) Front base width of LV-Atx3Q72 (C) and of Tg-Q69 model (D). Beclin 1-treated mice have a narrower front base width compared to control-treated mice. Values are expressed as mean ± SEM (n = 8; **P < 0.01; ***P < 0.001). (E and F) Hind base width of LV-Atx3Q72 (E) and of Tg-Q69 model (F). No statistical differences were observed between the LV model and wild-type (E) and no statistical differences were observed between control and beclin 1-treated Tg mice (F). Values are expressed as mean ± SEM (n = 8; *P < 0.05; n.s. = no significant difference). (G and H) Distance between the placement of hind and front paw in consecutive steps or footprint overlap. In both models beclin 1-treated mice are statistically different from control-treated and no different from wild-type mice regarding the distance of footprint overlap. Values are expressed as mean ± SEM (n = 8; *P < 0.05; **P < 0.01; ***P < 0.001).
Surprisingly, already at T0 the Tg-Q69 mice were phenotypically distinct from the wild-type regarding gait (reaching statistical difference on the stride length, Fig. 4B), beclin 1 was able to significantly increase the stride length (Fig. 4B, n = 8; 6.50 ± 0.26 versus 5.56 ± 0.21 in control, P < 0.01) and decrease the distance between front and hind limb overlap (Fig. 4H, n = 8; 0.73 ± 0.07 versus 1.17 ± 0.16 in control, P < 0.05) at 10 weeks post-injection. No significant differences were reached regarding the front base width (Fig. 4D) and hind base width (Fig. 4F) even though beclin 1 mediated a slight tendency for improvement. Importantly, at that time-point beclin 1-expressing Tg-Q69 mice were not statistically different from wild-type concerning the footprint overlap measurement (Fig. 4H). These results show that beclin 1 is able not only to hamper the development of an ataxic gait when expressed before onset of disease (LV-Atx3Q72), but also to mediate a partial rescue when administered after onset of symptoms in a severe-progressive model of polyglutamine disease/SCA (Tg-Q69).
Beclin 1 decreases aggregation of polyglutamine–expanded ataxin 3
Machado-Joseph disease as well as the other polyglutamine diseases is characterized by the presence of insoluble aggregates of expanded protein, in this case of ataxin 3 (Paulson et al., 1997). In previous work we showed that beclin 1 was able to increase the autophagic flux, consequently leading to an improved clearance of mutant ataxin 3 protein and decrease the number of mutant aggregates present in the rat striatum (Nascimento-Ferreira et al., 2011). Therefore in the present work we investigated if beclin 1 would clear mutant ataxin 3 aggregates formed in the cerebellum of LV-Atx3Q72 and Tg-Q69 mice (Fig. 5). Because the aggregates formed by both of these models had been previously characterized (Torashima et al., 2008; Nobrega et al., 2012) here we only focused on the clearance quantification of ataxin 3 aggregates stained with the 1H9 antibody (Fig. 5A–C) and haemagglutinin (Fig. 5D–H). In the LV-Atx3Q72 model, at 10 weeks post-injection, beclin 1 mediated a robust clearance of mutant ataxin 3 aggregates relative to controls (Fig. 5A–C; 1H9 immunostaining; n = 6; 282.6 ± 57.92 versus 926.3 ± 172.5 in control, P = 0.0054). Interestingly, at that same time-point the pattern of truncated ataxin 3 staining in Tg-Q69 mice was mainly diffuse with very few aggregates (Fig. 5D–H; haemagglutinin tag immunostaining). When present, aggregates were mainly present in the Purkinje cells of control-injected mice (Fig. 5D and F) (Fig. 5H; n = 4-5; 25.17 ± 2.275 versus 15.89 ± 2.241 in beclin 1-treated, P = 0.0244). These data show that beclin 1 expression decreases the level of polyglutamine-expanded ataxin 3 aggregates.
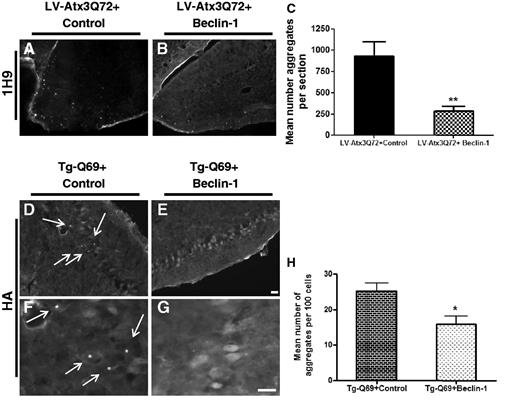
Beclin–1 alters the aggregation pattern. (A–C) Fluorescence microscopy analysis of mutant ataxin 3 aggregates in the lentiviral-based model (LV-Atx3Q72) revealed by the 1H9 antibody at 10 weeks post-injection. Aggregates were mainly present in the molecular and Purkinje cell layer of the cerebellar cortex and rarely seen in the granular layer. Scale bar = 20 µm. Quantification of mean number of aggregates per section revealed a beclin 1-mediated reduction of the number of aggregates as compared to control (C). Values are expressed as mean ± SEM (n = 6; **P < 0.01). (D–H) Fluorescence microscopy analysis of haemagglutinin (HA)-tagged truncated ataxin 3 protein in the transgenic model (Tg-Q69) at 10 weeks post-injection. Immunohistochemistry for haemagglutinin antibody revealed a predominantly diffuse expression of the mutant ataxin 3 protein. Aggregates were present occasionally mainly in Purkinje cells of the control-injected mice (D and F, arrows). Scale bar = 20 µm. Quantification of mean number of aggregates per 100 cells revealed a beclin 1-mediated reduction of the number of aggregates as compared to control (H). Values are expressed as mean ± SEM (n = 4–5; *P < 0.05).
Beclin 1 prevents neuronal dysfunction and neurodegeneration
To investigate whether impairments in motor function would correlate with histological changes in the cerebellum, we next examined the immunoreactivity for the dopamine- and cyclic AMP-regulated phosphoprotein of 32 KDa/DARPP-32 (now known as Ppp1r1b) and calbindin protein within the molecular and Purkinje cell layers. Both markers were present in Purkinje cell’s bodies and dendrites with similar staining patterns. Importantly, in the control-treated LV-Atx3Q72 model (Fig. 6A), reduced dendritic arborizations of the Purkinje cells and decreased immunoreactivity for DARPP-32 at the level of Purkinje cells and dendrites were observed, compared with beclin 1-treated mice (Fig. 6B) and wild-type (Fig. 6C). By quantifying the DARPP-32 immunoreactivity by optical densitometry (Fig. 6G) we observed that not only beclin 1-treated mice had an increased immunoreactivity for DARPP-32 compared with control-treated, but also were very similar to wild-type mice (Fig. 6G; n = 6; 3.02 ± 0.21 in control versus 6.96 ± 1.24 in beclin 1-treated; P < 0.05; wild-type: 7.76 ± 1.05). Similar findings were observed using the calbindin marker (Fig. 6D–F and H). In addition, no differences were observed in the molecular layer thickness in this model when compared with wild-type mice (Fig. 6I), which indicates that cerebellar atrophy was not present in this model at this 10 weeks time-point.
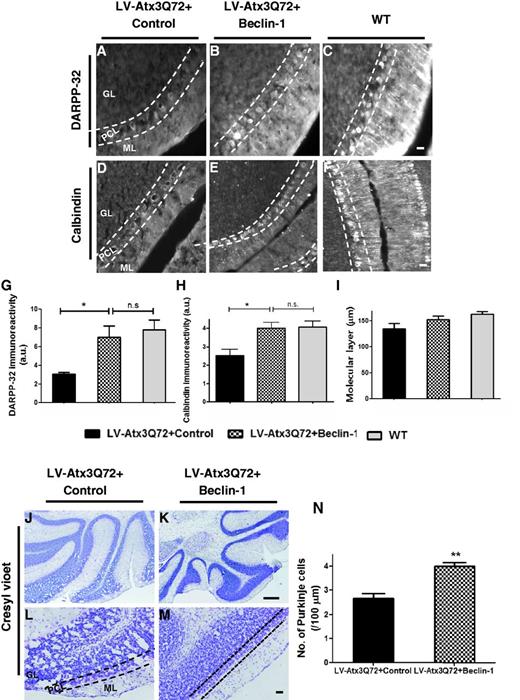
Beclin–1 protects from neuronal dysfunction. (A–C) Fluorescence microscopy analysis for the DARPP-32 protein in the lentiviral model (LV-Atx3Q72) and wild-type mice (WT). Immunohistochemistry for the DARPP-32 protein highlighted the Purkinje cells and their dendritic projection to the molecular layer. An increased expression of DARPP-32 neuronal marker was observed for the beclin 1-treated mice (B) relative to control-treated (A). Scale bar = 20 µm. (G) Quantification of optical densitometry of DARPP-32 immunoreactivity. Beclin 1 significantly increased DARPP-32 expression as compared to control. Note that beclin 1-injected mice are not statistically different than wild-type mice regarding the DARPP-32 immunoreactivity. Values are expressed as mean ± SEM (n = 6; *P < 0.05; n.s = no significant difference). (D–F) Fluorescence microscopy analysis for calbindin protein in the lentiviral model (LV-Atx3Q72) and wild-type mice (WT). An increased expression of calbindin marker was observed for the beclin 1-treated mice (E) relative to control-treated (D). Scale bar = 20 µm. (H) Quantification of optical densitometry of calbindin immunoreactivity. Beclin 1 significantly increased calbindin expression as compared to control. Values are expressed as mean ± SEM (n = 4; *P < 0.05; n.s. = no significant difference). (I) Quantification of the width of the molecular layer of the cerebellum by the DARPP-32 staining. No significant differences were observed between the lentiviral-based model and wild-type mice. (J–N) Cresyl violet analysis in the lentiviral-based model. Cresyl violet staining in beclin 1-treated mice (K and M) highlighted a better cellular morphology and cell number as compared to control-treated mice (J and L). In the control-treated LV-Atx3Q72 mice (J and L) a vacuolar pattern were present in the granular and Purkinje cell layer. Scale bar = 200 µm (J and K) and 40 µm (L and M). (N) Quantification of the Purkinje cell number. Beclin 1 overexpression protected mice from cell loss. Results are expressed as mean ± SEM (n = 4; **P < 0.001). ML = molecular layer; PCL = Purkinje cell layer; GL = granular layer.
To further examine morphological abnormalities between beclin 1 and control-treated LV-Atx3Q72 mice, a cresyl violet staining was performed (Fig. 6J–N). Beclin 1-injected mice revealed a preserved cellular morphology (Fig. 6K and M) as compared with control-treated mice (Fig. 6J and L), where several vacuoles were present in the granular and Purkinje cell layer. By quantifying the number of Purkinje cells per length unit, we observed a decreased cell number in the control-treated as compared to beclin-1 treated mice (Fig. 6N; n = 4; 4.00 ± 0.15 versus 2.67 ± 0.19 in control; P < 0.01).
Next, by analysing the Tg-Q69 model with the calbindin (Fig. 7A–C and J), DARPP-32 (Fig. 7D–F and K) and cresyl violet marker (Fig. 7G–I and L) we observed that beclin 1 overexpression mediated preservation of Purkinje cell morphology (Fig. 7B), dendritic arborizations (Fig. 7B, ML) and calbindin immunoreactivity (Fig. 7J) as compared with control-treated mice (Fig. 7A and J). No major differences were observed with the DARPP-32 staining (Fig. 7D–F and K). Interestingly, severe atrophy of the molecular layer of the cerebellum was apparent in this model as compared to wild-type mice (Fig. 7A–C and G–I, arrows), which was partially prevented by beclin 1 administration (Fig. 7B and H). Quantification of the molecular layer thickness confirmed that cerebellar atrophy was present in this model (Fig. 7L; n = 7; 23.34–29.86 µm in Tg-Q69 versus 156.5–164.7 µm in wild-type), which was hindered by beclin 1 overexpression (Fig. 7L; n = 7; 28.87 ± 0.99 in beclin 1-treated versus 24.30 ± 0.96 in control-treated mice; P < 0.01). In summary, these data show that beclin 1 prevents neuronal dysfunction and neurodegeneration, in agreement with the improvements observed in motor function.
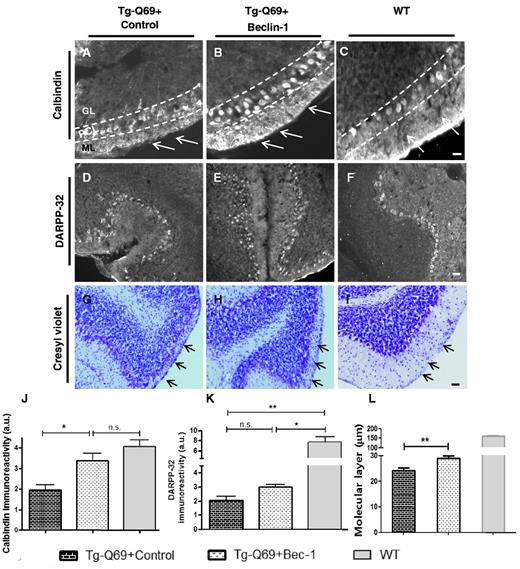
Beclin–1 protects against cerebellar atrophy. (A–C) Fluorescence microscopy analysis for the calbindin protein, in transgenic (A and B) and wild-type mice (C). Note the improved Purkinje cell morphology (B, PCL) and dendritic arborizations to the molecular layer (B, arrows) upon beclin 1 overexpression as compared to control (A). Scale bar = 40 µm. ML = molecular layer; PCL = Purkinje cell layer; GL = granular layer. (J) Quantification of optical densitometry of calbindin immunoreactivity. Beclin 1 significantly increased calbindin expression as compared to control, reaching similar values to wild-type mice. Values are expressed as mean ± SEM (n = 4; *P < 0.05; n.s. = no significant difference). (D–F) Fluorescence microscopy analysis for the DARPP-32 protein, in transgenic (D and E) and wild-type mice (F). (K) Quantification of optical densitometry of DARPP-32 immunoreactivity. No differences were observed between beclin 1 and control for this marker. Values are expressed as mean ± SEM (n = 3; *P < 0.05; **P < 0.01; n.s = no significant difference). (G–I) Cresyl violet staining. Note the reduced molecular layer width of the transgenic model (G and H, arrows) as compared to wild-type mice (I, arrows). Nevertheless, an increased width of the molecular layer in beclin 1-treated mice (H, arrows) relative to control-treated (G, arrows) was observed. Scale bar = 40 µm. (L) Quantification of the width of the molecular layer of the cerebellum by the calbindin staining. A significant increased molecular layer width was observed in the Tg-Q69 mice upon beclin 1 overexpression. Values are expressed as mean ± SEM (n = 7; **P < 0.001).
Discussion
Using both a presymptomatic lentiviral model and a severely affected transgenic mice modelling Machado–Joseph disease, we provide evidence that beclin 1 overexpression is able to prevent or partially rescue motor impairments characteristic of this disorder.
The simultaneous overexpression of beclin 1 and induction of Machado–Joseph disease (lentiviral-based model) was able to hamper the disease progression regarding balance, motor coordination and gait performances. After 10 weeks of beclin 1 overexpression, lentiviral-based Machado–Joseph disease mice (Nobrega et al., 2012) were indistinguishable from wild-type mice regarding the motor phenotype. These results are in agreement to previous reports in conditional mouse models of different polyglutamine diseases (Yamamoto et al., 2000; Zu et al., 2004; Boy et al., 2009). In these conditional mouse models the reversibility of the pathology was achieved by switching off the mutant protein transgene in an early stage of disease. In the present study, the mutant ataxin 3 transgene was continuously expressed, mimicking a more realistic scenario. This indicates that neural cells are capable of overcoming the pathology progression if protein clearance mechanisms remain functional.
Protein clearance mechanisms are particularly important for neurons, which can not eliminate their abnormal proteins or other toxic species by cell division (Cuervo, 2004; Wong and Cuervo, 2010). As insoluble polyglutamine inclusions are scarcely removed by the ubiquitin-proteasome system, which may become progressively impaired in these diseases (Bence et al., 2001; Venkatraman et al., 2004), autophagy is most probably responsible for the ability of these cells to clear mutant proteins and reverse the pathology after switching- off of the transgene in conditional disease models. Autophagy has been shown to play a central role in misfolded protein clearance within the CNS, particularly in diseases caused by insoluble and aggregation-prone proteins, such as Alzheimer’s disease, Parkinson’s disease and polyglutamine diseases (Ravikumar et al., 2004; Pickford et al., 2008; Spencer et al., 2009). In a previous study, we observed that autophagy was impaired in Machado–Joseph disease and beclin 1 levels decreased, whereas overexpression of beclin 1 stimulated the autophagy pathway, cleared misfolded ataxin 3 protein and protein aggregates while promoting neuroprotection, observed at the histological level.
Here, our main focus was to investigate beclin 1 effects on motor behaviour. Beclin 1-mediated neuroprotective effects were dependent on the timing of intervention and disease status. When administered to a transgenic mouse model with severe cerebellar atrophy and ataxic phenotype (Torashima et al., 2008), the observed effects, within the 10 week time-frame of the study, were relatively modest considering the results obtained with the lentiviral-based Machado–Joseph disease mice (Nobrega et al., 2012), which were indistinguishable from normal wild-type mice for most of the parameters studied. Nevertheless, beclin 1-overexpression in the transgenic mice mimicking a late stage of disease at the moment of viral injection in the cerebellum, led to a partial rescue of the phenotype, at the balance, motor coordination and gait levels. In addition, even though lentiviral vectors efficiently transduce Purkinje cells, cells targeted by the truncated ataxin 3 vector driven by the L7-promoter in this transgenic model, not all cells can be transduced by lentiviral vectors and therefore we can not exclude an insufficient beclin 1 expression within the whole cerebellum. Moreover, the duration of the study was the same used for the lentiviral-based model (10 weeks), which may not have been sufficient to maximize the therapeutic effects in the transgenic model. Therefore, beclin 1 effects on a middle stage of polyglutamine ataxia and in a late stage during longer periods of time remain to be investigated. Nevertheless, the results presented here are in agreement with the study involving a conditional mouse model of SCA1 (Zu et al., 2004), where complete reversibility of the disease was achieved by switching off the transgene in early and middle stages of disease, whereas the switching off in a severe stage improved the phenotype to a middle stage of disease.
Regarding neuropathology, in accordance with the motor behaviour we observed that an early beclin 1 expression was able to protect neuronal cells and impede the development of neuropathology evaluated by the number of cells with aggregates, DARPP-32 and calbindin immunoreactivity and number of Purkinje cells, whereas a ‘late’ administration could ameliorate the neuropathology but not completely prevent the cell damage, especially as when beclin 1 was administered to the transgenic mouse model, cerebellar atrophy had already taken place. Importantly, beclin 1 expression in the transgenic model was able to preserve Purkinje cell dendritic arborizations and the width of the molecular layer of cerebellar cortex, which was substantially decreased in control-treated compared with wild-type mice, indicative of cerebellar atrophy. The fact that beclin 1 was able to decrease the progression of cerebellar atrophy, revealed by the increased molecular layer width of treated animals is of major importance and indicative that even in a late stage of disease and despite the continuous expression of mutant transgene, it is possible to slow-down the progression of neurodegeneration and cell death.
The overall results obtained with beclin 1 overexpression in the lentiviral-based and transgenic mouse model relative to motor function and Purkinje cell pathology show a correlation between neuropathology and motor behaviour. Moreover these results indicate that Purkinje neurons retain the ability to recover from polyglutamine-expanded ataxin 3-induced toxicity, being able to improve complex motor skills such as motor coordination, balance and gait.
This work, despite overall supporting an early intervention for Machado-Joseph disease and polyglutamine diseases, shows that rescue of the pathology is also possible in advanced stages, at least partially, by upregulating beclin 1 levels. This effect was observed both for full-length and truncated mutant ataxin 3 and may also benefit other polyglutamine diseases.
Machado–Joseph disease as well as other polyglutamine diseases is a genetic hereditary disease with known aetiology. Presently, genetic testing to diagnose Machado–Joseph disease can be performed pre-implantation, in uterus or after birth. This indicates that once available, a therapeutic treatment can be initiated before onset of symptoms. In this regard, the knowledge that upregulation of beclin 1 is able to clear the initial disease-causing mutant ataxin 3 protein and delay the progression of motor impairments may be of major importance. Moreover, it has been recently shown that some drugs upregulate beclin 1 (Can et al., 2011; Huang et al., 2011), introducing new therapeutic value for these drugs and hopefully for several neurodegenerative diseases. Unfortunately, given that these drugs may account for several secondary effects, no perfect drug or treatment is for the moment available for Machado–Joseph disease.
Altogether this study shows that overexpression of beclin 1 is able to prevent the polyglutamine-related behaviour and neuropathology, when administered to an early stage and to partially hamper the progression of the pathological phenotype when administered to a late stage of the disease. As beclin 1 has been found to be decreased in neurodegenerative diseases; upregulation of beclin 1, genetically or pharmacologically may represent a promising therapeutic approach for polyglutamine diseases and overall to neurodegenerative diseases.
Acknowledgements
We thank Dr. Lígia Sousa-Ferreira and Dr. Luísa Cortes for the assistance with the confocal microscopy and Prof. Beth Levine for the beclin 1 construct that we used for subcloning into the lentiviral backbone.
Funding
This work was supported by the Portuguese Foundation for Science and Technology (PTDC/SAU-NEU/099307/2008 and PEst-C/SAU/LA0001/2011), the National Ataxia Foundation (Research Award 2010), the Association Française pour les Myopathies (SB/NF/2010/2008 Number 15079CA), and The Richard Chin and Lily Lock Machado-Joseph disease Research Fund. INF, CN, CA, DA and IO were also supported by the Portuguese Foundation for Science and Technology (fellowships: SFRH/BD/29479/2006, SFRH/BPD/73942/2010, SFRH/BPD/62945/2009, SFRH/BD/61461/2009).
Abbreviation
References
Author notes
*These authors contributed equally to this work


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}