




Abstract
Upregulation of microRNA-21 (miR-21) is known to be strongly associated with the proliferation, invasion, and radio-resistance of glioma cells. However, the regulatory mechanism that governs the biogenesis of miR-21 in glioma is still unclear. Here, we demonstrate that the DEAD-box RNA helicase, DDX23, promotes miR-21 biogenesis at the post-transcriptional level. The expression of DDX23 was enhanced in glioma tissues compared to normal brain, and expression level of DDX23 was highly associated with poor survival of glioma patients. Specific knockdown of DDX23 expression suppressed glioma cell proliferation and invasion in vitro and in vivo, which is similar to the function of miR-21. We found that DDX23 increased the level of miR-21 by promoting primary-to-precursor processing of miR-21 through an interaction with the Drosha microprocessor. Mutagenesis experiments critically demonstrated that the helicase activity of DDX23 was essential for the processing (cropping) of miR-21, and we further found that ivermectin, a RNA helicase inhibitor, decreased miR-21 levels by potentially inhibiting DDX23 activity and blocked invasion and cell proliferation. Moreover, treatment of ivermectin decreased glioma growth in mouse xenografts. Taken together, these results suggest that DDX23 plays an essential role in glioma progression, and might thus be a potential novel target for the therapeutic treatment of glioma.
Upregulation of microRNA-21 (miR-21) is strongly associated with glioma malignancy, but the regulatory mechanism that governs miR-21 biogenesis is unclear. Yin et al. demonstrate that the DEAD-box RNA helicase DDX23 promotes miR-21 biogenesis at the post-transcriptional level in glioma cells, and that DDX23 inhibition reduces glioma growth in mouse xenografts.
Introduction
MicroRNA-21 [miR-21; miRBase (http://www.mirbase.org) accession number: MI0000077] is known to be overexpressed in various cancers, and we previously reported that miR-21 overexpression in glioma was associated with glioma cell invasion, proliferation and radio-resistance (Kwak et al., 2011; Gwak et al., 2012). Interestingly, miR-21 expression was shown to be post-transcriptionally upregulated by growth factors, hyaluronic acid and radiation (Kim et al., 2011; Kwak et al., 2011; Gwak et al., 2012). However, the regulatory mechanism governing miR-21 biogenesis is still unclear.
One of the key regulatory steps in miRNA biogenesis is the primary-to-precursor transcript turnover step (cropping step), which is conducted by the Drosha microprocessor (Kim et al., 2009; Ha and Kim, 2014). Drosha is a key regulatory RNase that generates a ∼65-nucleotide stem-and-loop structure from the primary miRNA (pri-miRNA) transcript (Kim et al., 2009; Ha and Kim, 2014). Additional and miRNA subgroup-specific auxiliary factors also contribute to the cropping step. Recently, RNA regulatory proteins, such as hnRNP A1 (encoded by HNRNPA1) (Guil and Caceres, 2007) and KSRP (encoded by KHSRP) (Trabucchi et al., 2009), have been shown to directly bind to pri-miRNAs and facilitate Drosha processing. Moreover, we recently reported that RNH1 interacts specifically with Drosha and pri-miR-21 together (Kim et al., 2011).
Members of the DEAD-box family of RNA helicases play important roles in various aspects of RNA processing, including transcription, spliceosome biogenesis, ribosome biogenesis, splicing, nucleocytoplasmic transport, translation and decay (Abdelhaleem, 2004; Fuller-Pace, 2013). These family members share a conserved core that includes the amino acid sequence, D-E-A-D (aspartate-glutamate-alanine-aspartate). They use energy received from ATP hydrolysis to unwind double-stranded RNA, generally act as components of multi-protein complexes, and have diverse functions that depend on their interacting partners. However, the specific functions of most DEAD-box RNA helicases are not yet known.
Several DEAD-box RNA helicases are aberrantly expressed in various types of cancer, where they may play important roles in cancer development and/or progression (Abdelhaleem, 2004; Fuller-Pace, 2013). For example, DDX1 overexpression is a potential prognostic marker for early recurrence in breast cancer (Germain et al., 2011), and DDX1 knockdown in testicular tumour cells abolishes their ability to form tumours in nude mice (Tanaka et al., 2009). Similarly, DDX3 is overexpressed in hepatocellular carcinoma, and the overexpression of DDX3 in liver cancer cell lines has been associated with cellular transformation (Huang et al., 2004). The gene encoding DDX5 is amplified in breast cancer, and DDX5 knockdown impairs the proliferation of breast cancer cells (Mazurek et al., 2012). Finally, overexpression of DDX6 is found in several cancers, and DDX6 knockdown inhibits the tumour growth of colorectal cancer cells in nude mice (Akao et al., 1995). Despite the advances made by these previous studies, most of them failed, however, to elucidate the mechanism(s) through which the DDXs affect cancer progression (Lin et al., 2008).
The DEAD-box RNA helicase, DDX23 (also known as PRP28), acts as a component of the U4/U6-U5 tri-snRNP complex involved in pre-mRNA splicing, and is necessary for the formation of the spliceosomal B complex (Mathew et al., 2008). However, researchers do not fully understand yet the specific role of DDX23 in cancer, especially glioma.
In this study, we found that DDX23 was upregulated in glioma, where its expression level was associated with patient survival. Moreover, specific knockdown or overexpression of DDX23 was found to modulate glioma cell proliferation and invasion both in vitro and in vivo, in conjunction with the Drosha microprocessor, by regulating the cropping step during the biogenesis of the well-known oncogenic miRNA, miR-21.
Materials and methods
Cell culture and reagents
Human embryonic kidney cell line 293T, human glioma cell line U87MG and human cervical cancer cell line HeLa were obtained from the American Type Culture Collection. The 293FT cell line was purchased from Life Technologies. Human non-small cell lung cancer cell line A549, human prostate cancer cell line DU145 and pancreatic cancer cell line PANC-1 were obtained from Kyungsil Yoon (National Cancer Center, Republic of Korea), Sang-Jin Lee (National Cancer Center, Republic of Korea), and Yun-Hee Kim (National Cancer Center, Republic of Korea), respectively. 293T, U87MG, HeLa and PANC-1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM, HyClone). A549 and DU145 cells were cultured in RPMI-1640 media (HyClone). All media were supplemented with 10% foetal bovine serum (FBS) (HyClone), penicillin/streptomycin (Welgene) and 10 µg/ml ciprofloxacin (Santa Cruz Biotech). Two glioma patient-derived primary cultures (X01 and CSC2; Jin et al., 2012; Yin et al., 2014) were maintained in DMEM/F-12 supplemented with B27 (Life Technologies), epidermal growth factor (EGF, 20 ng/ml; R&D Systems) and basic fibroblast growth factor (bFGF, 20 ng/ml; R&D Systems).
Invasion and proliferation assays
Invasion assays were performed using modified Boyden chambers with polycarbonate nucleopore membranes (Corning) as previously reported (Yin et al., 2011). Briefly, 3 × 104 cells in serum-free medium were placed into the upper chamber of an insert coated with Matrigel® (BD Biosciences). Medium containing 10% foetal bovine serum was added to the lower chamber. After incubation at 37°C for 24 h, the cells remaining on the upper membrane were removed with cotton wool, whereas the cells that had invaded through the membrane were stained with Diff-Quick (Microptic). Invasiveness was determined by counting cells in five microscopic fields per well, and the extent of invasion was expressed as the average number of cells per microscopic field. Proliferation assays were performed by using the CyQUANT® NF cell proliferation assay kit (Life Technologies) according to the manufacturer's protocol.
Plasmids
For the generation of pEGFPc1-DDX23, plasmid containing the full-length open reading frame of human DDX23 (hMU005548; 21C Frontier Human Gene Bank, Republic of Korea), was PCR-amplified with the following oligomers specific for DDX23: sense, 5′-GGAATTCTGTGATGGCAGGAGAGCTGG-3′ and antisense, 5′-CGGGATCCGAAGAGTGCTGTGTCAGGCAAAG-3′. BamHI-EcoRI digested PCR product was inserted into pEGFPc1 (Takara). pFLAG-CMV2-DDX23 was generated by ligation of BamHI-EcoRI treated pEGFPc1-DDX23, and BamHI-EcoRI-CIP treated pFLAG-CMV2 (Sigma-Aldrich) together. For the generation of pEGFPc1-DDX23(K441N), plasmid containing the full-length open reading frame of human DDX23 (pEGFPc1-DDX23) was PCR-amplified with the following oligomers specific for DDX23: N-terminal fragment; sense, 5′-GGAATTCTGTGATGGCAGGAGAGCTGG-3′ and antisense, 5′-GAGGAAGGCTGCTGTGTTGCCACTGCCAGTCTC-3′; C-terminal fragment; sense, 5′-GAGACTGGCAGTGGCAACACAGCAGCCTTCCTC-3′ and antisense, 5′-CGGGATCCGAAGAGTGCTGTGTCAGGCAAAG-3′; DpnI digested PCR products were assessed for the additional PCR cycle with full-length DDX23 specific primers sense, 5′-GGAATTCTGTGATGGCAGGAGAGCTGG-3′ and antisense, 5′-CGGGATCCGAAGAGTGCTGTGTCAGGCAAAG-3′; ∼2.3-kb PCR product was digested with BamHI-EcoRI and inserted into BamHI-EcoRI-CIP treated pEGFPc1. For the generation of HRST-FLAG-DDX23(WT)-IRES-GFP and HRST-FLAG-DDX23(K441N)-IRES-GFP constructs for the lentiviral transduction, PCR was performed with pEGFPc1-DDX23(WT) and pEGFPc1-DDX23(K441N) with the following oligomers; sense, 5′-TTTGCGGCCGCCATGGACTACAAAGACGATGACGACAAGATGGCAGGAGAGCTGGC-3′ and antisense, 5′-CGGGATCCGAAGAGTGCTGTGTCAGGCAAAG-3′, respectively. The amplified DNA fragments were digested with NotI-BamHI and subcloned into HRST-IRES-GFP (Yin et al., 2014) treated with NotI-BamHI-CIP. For the generation four consecutive miR-21 target sites bearing sensor at the firefly 3′ end, complementary oligonucleotides (sense, 5′-TCGAGTCAACATCAGTCTGATAAGCTATCAACATCAGTCTGATAAGCTATCAACATCAGTCTGATAAGCTATCAACATCAGTCTGATAAGCTAC-3′ and antisense, 5′-TCGAGTAGCTTATCAGACTGATGTTGATAGCTTATCAGACTGATGTTGATAGCTTATCAGACTGATGTTGATAGCTTATCAGACTGATGTTGAC-3′) were annealed, phosphorylated with T4 PNK (New England Biolabs), and then inserted into the pcDNA6.2-Fluc (Kim et al., 2011), which was digested with XhoI. pGIPZ non-silencing shRNAmir lentiviral control vector (pGIPZ-Con; RHS4346) and pGIPZ-shDDX23 (V3LHS_641139) were purchased from Open Biosystems. Construction procedures of pcDNA3-pri-miR-21, pri-miR-21 sensor, pCK-Drosha-FLAG(WT), pCK-Drosha-FLAG(TN), pCK-FLAG-Dicer(WT), pCK-FLAG-Dicer(TN), pcDNA3-FLAG-Lin28B(WT), pEGFPc1-His×6-KSRP are described elsewhere (Kim et al., 2011; Lee et al., 2014). All oligomers were purchased from Bioneer or Macrogen. All constructs were verified by DNA sequencing (Cosmo Genetech).
Small interfering RNAs and transient transfection
Small interfering RNAs (siRNAs) were synthesized by Bioneer. siRNA for human DDX23 (siDDX23; 5′-CCUGUGCAAGUUUGGUGCU-3′) and as a negative control, scrambled siRNA (siCon; 5′-GUUCAGCGUGUCCGGCGAG-3′) were used. Cells (106) were plated on 100 mm culture dish 24 h before transfection. Five hundred picomoles of siRNA and 15 μl of Lipofectamine™ 2000 (Life Technologies) were used for each transient transfection for northern blotting, invasion and proliferation assays.
Lentivirus production and infection
293FT cells (3–4 × 106) (Life Technologies) were plated on 100 mm culture dish 24 h before transfection. Lentiviral construct (4.5 μg) [HRST-FLAG-DDX23(WT)-IRES-GFP, HRST-FLAG-DDX23(K441N)-IRES-GFP, pGIPZ-Con, or pGIPZ-shDDX23], 3 μg of psPAX2 (Addgene; #12260), and 1.5 μg of pMD2.G (Addgene; #12259) were co-transfected into 293FT cells using 27 μl of Lipofectamine™ 2000 (Life Technologies) or METAFECTENE® PRO (Biontex). The Opti-MEM® medium (Life Technologies) containing transfectant was changed complete medium without antibiotics 5 h after transfection. The medium containing lentivirus was harvested at 48–72 h after transfection. Lentivirus containing medium directly used for the each infection or lentiviral particles was concentrated and purified using a Lenti-X™ concentrator (Takara). Cells were infected with lentivirus in the presence of 10 μg/ml polybrene (Sigma-Aldrich).
Antibodies and western blotting
Anti-DDX23 (rabbit polyclonal, 1/500 dilution, Bethyl Laboratories), anti-β-actin (clone C4, mouse monoclonal, 1/500 dilution, Merck Millipore), anti-α-tubulin (TU-02, mouse monoclonal, 1/2000 dilution, Santa Cruz Biotech), anti-GAPDH (14C10, rabbit monoclonal, 1/1000 dilution, Cell Signaling), anti-Myc (A-14, rabbit polyclonal, 1/1000 dilution, Santa Cruz Biotech), anti-FLAG (clone M2, mouse monoclonal, 1/2000 dilution, Sigma-Aldrich), anti-DYKDDDDK (clone L5, rat monoclonal, 1/2000 dilution, BioLegend), anti-Drosha (rabbit polyclonal, 1/2000 dilution, Abcam), anti-hnRNP A1 (clone 4B10, mouse monoclonal, 1/2000 dilution) (Choi et al., 2014; Lee et al., 2014), anti-eIF4GI (anti-4GI; rabbit polyclonal, 1/5000 dilution) (Lee et al., 2014), anti-HuR (clone 3A2, mouse monoclonal, 1/1000 dilution, Santa Cruz Biotech), and anti-GFP (clone B2, mouse monoclonal 1/2000 dilution, Santa Cruz Biotech) antibodies were used through the all western blot analyses. As a secondary antibody, horseradish peroxidase-conjugated anti-rabbit (1/5000 dilution, Vector Laboratories), anti-mouse (1/5000 dilution, Vector Laboratories), and anti-rat immunoglobulin (1/5000 dilution, Santa Cruz Biotech) were used.
Small RNA analysis by northern blotting
Total RNAs were isolated with TRIzol® (Life Technologies) and northern blotting was carried out N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC; Sigma-Aldrich) cross-linking method (Lee et al., 2014). In brief, 1–10 µg of total RNA was separated on 12% SequaGel (National Diagnostics) and transferred to Hybond NX nylon membrane (GE Healthcare Life Sciences). The membrane was hybridized overnight at 42°C with 32P 5′-end-labelled oligonucleotide probes (anti-miR-21; 5′-TCAACATCAGTCTGATAAGCTA-3′, anti-let-7a; 5′-AACTATACAACCTACTACCTCA-3′ and anti-U6; 5′-GCTTCACGAATTTGCGTGTCATCCT-3′) in PerfectHyb Plus hybridization buffer (Sigma-Aldrich) and then washed according to standard procedures. Radioactive signals were scanned by the BAS-2500 analyzer (Fujifilm) or obtained from X-ray film (AGFA) exposure at −80°C.
Immunoprecipitation, RNP-immunoprecipitation and semi-quantitative RT-PCR
Immunoprecipitation was performed as previously described (Kim and Richter, 2006, 2007; Choi et al., 2014; Lee et al., 2014; Yin et al., 2014). Cells were crushed in immunoprecipitation buffer [150 mM NaCl, 25 mM HEPES-KOH (pH 7.5), 10% (v/v) glycerol, 1 mM MgCl2, 2 mM sodium orthovanadate, 2 mM β-glycerophosphate, 1 mM phenylmethylsulphonyl fluoride, 1 mM dithiothreitol, 2 mM EDTA, 0.5% Triton™ X-100 and 1 × protease inhibitor cocktail (Roche Applied Science)]. After brief sonication and incubation on ice, lysates were centrifuged at 12 000g for 5 min to remove insoluble materials. The lysates were then incubated with anti-FLAG M2 affinity gels (Sigma-Aldrich) for 2 h at 4°C. The collected beads were then washed four times with washing buffer (0.05% Triton™ X-100 immunoprecipitation buffer without protease inhibitor cocktail) and boiled in SDS sample buffer for western blot analysis. Ribonucleoprotein (RNP) immunoprecipitation was performed by the same strategy as protein immunoprecipitation except RNaseOUT™ (Life Technologies) was included in the reaction mixture. The lysates were then incubated with anti-FLAG M2 affinity gels for 1 h at 4°C. The collected beads were then washed four times with washing buffer and the total RNA was isolated using TRIzol® (Life Technologies) according to the manufacturer’s instructions. cDNAs were synthesized using ImProm-II™ reverse transcription system (Promega) with oligo(dT)20 (Bioneer) according to the manufacturer’s instructions. Reaction parameters are as followed; 25–30 cycles of 95°C for 20 s, 55°C for 40 s, and 72°C for 20 s. The PCRs were performed with specific oligomers (Bioneer) for pri-miR-21 (NR_029493.1), pri-miR-29a (NR_029503.1) and GAPDH with e-Taq (Solgent): pri-miR-21, 5′-CGGGATCCAAATCCTGCCTGACTGTCTGC-3′ and 5′-GGAATTCTGATTATAAACAATGATGCTGG-3′; pri-miR-29 a, 5′-CGGGATCCAAGAGCCCAATGTATGCTGG-3′ and 5′-GGAATTCAACGGTCACCAATACATTTCC-3′; GAPDH, 5′-GGAGTCCACTGGCGTCTTCAC-3′ and 5′-GAGGCATTGCTGATGATCTTGAGG-3′. In some case human β-actin (encoded by ACTB) was used as loading control. β-actin, 5′-CCTGGAACGGTGAAGGTGACA-3′ and 5′-AAGGGACTTCCTGTAACAATGCA-3′. The PCR products were analysed on the 1% agarose gel.
Luciferase assays for miR-21 sensors
Primary or mature miR-21 sensors were cotransfected with normalization control pRL-TK (Promega; Renilla luciferase) into 293T cells by using Lipofectamine™ 2000 (Life Technologies) or METAFECTENE® PRO (Biontex) according to the manufacturer’s instructions. After 48 h, cells were lysed with passive lysis buffer (Promega). Aliquots of lysates were analysed by Dual-Luciferase® Reporter Assay System (Promega). The sensor signal from the firefly luciferase was first normalized with that from Renilla (pRL-TK; Promega). Then the signal was renormalized with that from the firefly luciferase that lacked pri-miR-21 or miR-21 binding sites (control sensor).
Xenograft mouse models
All animal research was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at the National Cancer Center, Republic of Korea. All animals used were 6-week-old female Balb/c nude mice. Orthotopically transplanted brain tumour model was established by injecting 105 lentivirus-infected U87MG glioma cells into the mouse brain. Lentivirus-infected U87MG glioma cells were harvested, washed, resuspended in Dulbecco's phosphate-buffered saline, and then injected stereotactically into the left striatum of mouse brain. The injection coordinates were 2.2 mm to the left of the midline and 0.2 mm posterior to the bregma at a depth of 3.5 mm. After 4 weeks, MRI images were acquired. Subcutaneous xenograft model was established by injecting 5 × 106 U87MG glioma cells subcutaneously into the hip area on both sides of nude mice. Subcutaneous xenograft mice were administered with control vehicle [propylene glycol + glycerol formol (60:40 v/v)] or ivermectin (IVOMEC®, Merial) as an intratumoural route. Dosage of ivermectin was used by 3 mg/kg or 10 mg/kg. After 6 weeks, mice were sacrificed and subcutaneous xenograft tumours were isolated.
Magnetic resonance imaging
MRI analysis was performed and images were acquired using a 7.0 T magnet (BioSpin, Bruker). After localizer imaging on three orthogonal axes, T2-weighted images of the entire mouse brain were acquired using a Rapid Acquisition with Refocused Echoes (RARE) sequence with repetition time and echo time set to 2500 and 35 ms, respectively. Other parameters used were a 2 cm field of view and a 256 × 256 matrix in four averages, resulting in a total scan time of 4 min. Tumour mass was measured by Philips DICOM Viewer (Philips).
Histology, immunohistochemical staining and TUNEL assay
To allow observation of histological features, mice were anaesthetized with isoflurane and euthanized by transcardial perfusion with 10 ml of phosphate-buffered saline, followed by 10 ml of 4% paraformaldehyde solution. The brains or subcutaneous tumour tissues were isolated, fixed with 4% paraformaldehyde for 24 h at 4°C, and stained with haematoxylin (Dako) and 0.25% eosin (Merck Millipore). Immunohistochemical staining was performed with the automated instrument Discovery XT (Ventana Medical System) as follows; sections were deparaffinized and rehydrated with EZ Prep (Ventana Medical System) and washed with reaction buffer (Ventana Medical System). The antigens were retrieved with heat treatment in pH 8.0 Tris-EDTA buffer (CC1, Ventana Medical System) at 90°C for 30 min for anti-Cleaved caspase-3 (Cell Signaling, #9661, 1/500) and pH 6.0 citrate buffer (Ribo CC, Ventana Medical System) at 90°C for 30 min for anti-Ki67 (Abcam, #15580, 1/2000). Apoptotic U87MG glioma cells in subcutaneous xenograft tumour tissues were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) using ApopTag® Red In Situ Apoptosis Detection Kit (Merck Millipore).
REMBRANDT database analysis
DDX23 and miR-21 expression and survival data of glioma patients were obtained from the REMBRANDT (Repository of Molecular Brain Neoplasia Data) database of the National Cancer Institute, USA (http://caintegrator-info.nci.nih.gov/rembrandt). Kaplan-Meier survival plots were analysed by Statistical Package for the Social Sciences software version 12.0 (SPSS).
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) determined from a minimum of three independent experiments. Differences were assessed by the two-tailed Student's t-test using Excel software (Microsoft). ***P < 0.001, **P < 0.01, and *P < 0.05 were considered statistically significant.
Results
DDX23 expression is upregulated in glioma patients
To address functional association of DDX23 in glioma progression, we first analysed the DDX23 mRNA expression data found in the REMBRANDT database (Madhavan et al., 2009), and observed that the mRNA expression levels of DDX23 were significantly higher in tumour samples from 148 patients with astrocytoma, 67 with oligodendroglioma, 11 with mixed oligoastrocytoma, and 228 with glioblastoma multiforme, compared to 28 non-tumour brain tissue samples (Fig. 1A). Overall survival was significantly shorter in glioma patients with 4-fold higher DDX23 mRNA expression levels compared to patients with intermediate levels (Fig. 1B).
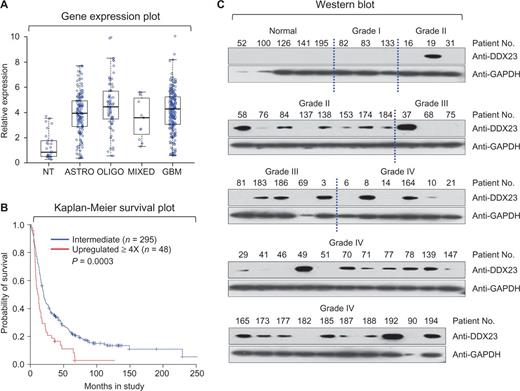
DDX23 expression is correlated with glioma progression and overall survival of glioma patients. (A) The expression level of mRNA obtained from non-tumour (NT, n = 28), astrocytoma (ASTRO, n = 148), oligodendroglioma (OLIGO, n = 67), mixed oligoastrocytoma (MIXED, n = 11), and glioblastoma multiforme (GBM, n = 228). Data obtained from the REMBRANDT database of the National Cancer Institute, USA. (B) Kaplan-Meier plot of survival for patients with DDX23 mRNA levels upregulated 4-fold is significantly higher than for all other brain tumour patients (DDX23 upregulated 4-fold, n = 48; DDX23 intermediate, n = 295; P = 0.0003) based on the log-rank test. Data obtained from the REMBRANDT database of the National Cancer Institute, USA. (C) Western blot of DDX23 using protein extracts from patient tissues from the National Cancer Center, Republic of Korea. GADPH was used as a loading control.
Next, we analysed the protein levels of DDX23 in tumour tissues of 49 glioma patients and five normal controls, which were obtained from the National Cancer Center, Republic of Korea. The DDX23 protein was not detected in normal brains and grade I glioma tissues, whereas significantly higher expression was found in grade II, III and IV glioma tissues (Fig. 1C). As grade I gliomas are known as benign tumours, whereas grade II, III and IV gliomas are characterized as malignant tumours (Furnari et al., 2007), these results suggest that upregulation of DDX23 mRNA and protein is strongly associated with malignancy of gliomas.
DDX23 is essential for the invasion and proliferation of glioma cells
To examine the function of DDX23 upregulation in glioma malignancy, we performed short hairpin (sh) RNA-mediated knockdown of DDX23 expression in the U87MG glioma cells (Fig. 2A). Invasion assays revealed that DDX23 knockdown reduced the highly invasive phenotype of U87MG glioma cells (Fig. 2A), suggesting that DDX23 is required for glioma invasion. Next, we overexpressed DDX23 in U87MG glioma cells via lentiviral transduction, and found that DDX23 overexpression increased glioma cell invasion (Supplementary Fig. 2A). Moreover, the proliferation of U87MG glioma cells was suppressed by DDX23 knockdown (Fig. 2B) but potentiated by DDX23 overexpression (Supplementary Fig. 2B). These data demonstrated that DDX23 is a critical regulator of glioma cell invasion and proliferation.
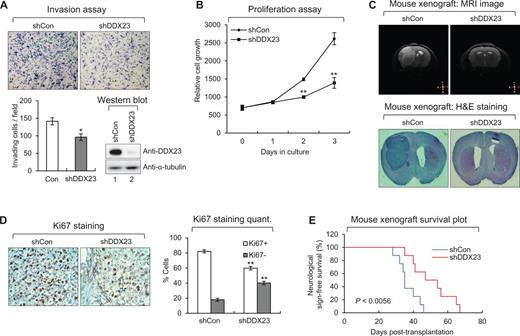
DDX23 promotes invasion and proliferation of U87MG glioma cells. (A) Invasion assay was performed in U87MG glioma cells infected with shDDX23 expressing lentiviral or control construct. Representative photos of invaded cells (top), quantitative data of invaded cells (left lower panel), and knockdown of DDX23 was confirmed by western blot (right lower panel). All error bars in graph represent mean ± SEM (n = 3). *P < 0.05. (B) Proliferation assay was performed in U87MG glioma cells infected with shDDX23 expressing lentiviral or control construct. All error bars in the graph represent mean ± SEM (n = 3). **P < 0.01. (C) Representative photos of MRI image (upper) and haematoxylin and eosin (H&E) staining (lower) in tumours derived from the orthotopically injected with U87MG-shCon and U87MG-shDDX23 cells. (D) Ki67 staining in tumours derived from the orthotopically injected with U87MG-shCon and U87MG-shDDX23 cells. Representative photos (left) and quantitative data (right) were shown. All error bars in graph represent mean ± SEM (n = 3). **P < 0.01. (E) Kaplan-Meier survival curves of mouse implanted with U87MG-shCon and U87MG-shDDX23 cells. P < 0.0056.
Next, we examined the tumorigenicity of DDX23 in vivo using a mouse xenograft model. DDX23-knockdown U87MG glioma cells were orthotopically injected into the brains of nude mice. MRI of living animals and haematoxylin and eosin staining and of tumour-bearing brain slices showed that the tumour masses of DDX23-knockdown xenografts were significantly decreased compared to those of control mice (Fig. 2C and Supplementary Fig. 3), suggesting that DDX23 knockdown also suppressed glioma cell proliferation in vivo. Ki67 (a cell proliferation marker) staining confirmed that glioma cell proliferation was decreased in DDX23-knockdown xenografts compared to controls (Fig. 2D). Moreover, overall survival was significantly increased in mice harbouring DDX23-knockdown xenografts compared to controls (Fig. 2E). Taken together, our data demonstrate that DDX23 is critical for glioma development.
DDX23 modulates the biogenesis of miR-21
As shown in Figs 1 and 2, the DDX23-related phenotypes of glioma patients and the tested U87MG glioma cell line were largely similar to the miR-21-mediated phenotypes, such as cell proliferation and invasion, previously observed by our group and others (Gabriely et al., 2008; Papagiannakopoulos et al., 2008; Kim et al., 2011; Kwak et al., 2011). We therefore hypothesized that DDX23 may regulate the biogenesis of miR-21 through a yet undiscovered mechanism. To test this possibility, we used small RNA northern blot analysis to examine the levels of the precursor and mature miR-21 in DDX23-knockdown U87MG glioma cells and DDX23-overexpressing 293T cells. 293T cells were used for the overexpression of DDX23 and pri-miR-21 RNA together because of low transfection efficiency of U87MG glioma cells. Consistent with our hypothesis, DDX23-knockdown cells showed reductions in the levels of both precursor and mature miR-21 (Fig. 3A), whereas DDX23 overexpression induced upregulation of both species (Fig. 3B).
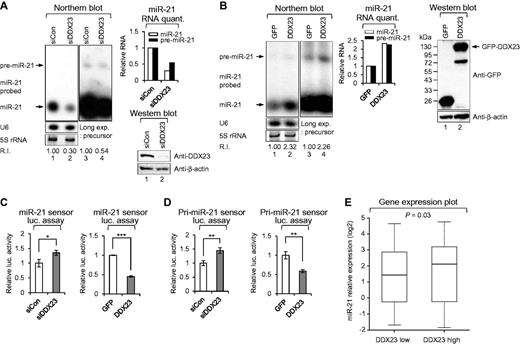
DDX23 modulates oncogenic miR-21 biogenesis at the post-transcriptional level. (A) Specific knockdown of DDX23 induced downregulation of miR-21. Scrambled (siCon) and DDX23 specific (siDDX23) siRNAs were transfected to U87MG glioma cells and then the level of endogenous miR-21 were monitored by northern blotting. After 48 h transfection, northern blot was performed with miR-21-specific 32P 5′-end-labelled oligonucleotide probe. Arrows indicate positions of precursor (upper) and mature (lower) miR-21. 5S rRNA was used as the loading control and U6 was used as hybridization control (lower left). The quantification of precursor and mature miR-21 are shown in graph (upper right). The experiments were repeated at least three times with similar results. The figure shown in A is representative. Protein level of DDX23 and β-actin (loading control) were confirmed by western blot after specific knockdown of DDX23 in U87MG glioma cells (lower right). R.I. = relative intensity. (B) Overexpression of DDX23 enhances the level of precursor and mature miR-21. Plasmids encoding GFP-DDX23 were cotransfected with pri-miR-21 RNA encoding plasmid into 293T cells. After 48 h transfection, northern blot was performed with miR-21-specific 32P 5′-end-labelled oligonucleotide probe. Arrows indicate positions of precursor (upper) and mature (lower) miR-21. 5S rRNA was used as the loading control and U6 was used as hybridization control (lower left). The quantification of precursor and mature miR-21 are shown in the graph (middle). The experiments were repeated at least three times with similar results. The figure shown in B is representative. Protein level of GFP-DDX23 and β-actin (loading control) were confirmed by western blot after overexpression of GFP-DDX23 in 293T cells (right). Long exp. = long exposure; R.I. = relative intensity. (C) The mature miR-21 sensor was cotransfected to 293T cells along with siRNA against DDX23 (siDDX23) or control siRNA (siCon) (left). Data represent the mean values of at least three independent experiments performed in triplicate (*P < 0.05). All error bars in graph represent mean ± SEM and the P-value compares the siCon to siDDX23. The mature miR-21 sensor was cotransfected to 293T cells along with GFP-DDX23 or control GFP-encoding plasmid (GFP) (right). Data represent the mean values of at least three independent experiments performed in triplicate (***P < 0.001). All error bars in graph represent mean ± SEM and the P-value compares GFP to GFP-DDX23. (D) The pri-miR-21 sensor was cotransfected to 293T cells along with siRNA against DDX23 (siDDX23) or control siRNA (siCon) (left). Data represent the mean values of at least three independent experiments performed in triplicate (**P < 0.01). All error bars in graph represent mean ± SEM and the P-value compares the siCon to siDDX23. The pri-miR-21 sensor was cotransfected to 293T cells along with GFP-DDX23 or control GFP-encoding plasmid (GFP) (right). Data represent the mean values of at least three independent experiments performed in triplicate (**P < 0.01). All error bars in graph represent mean ± SEM and the P-value compares the GFP to GFP-DDX23. (E) Relative expression levels of miR-21 in the DDX23-low and DDX23-high groups from the REMBRANDT database.
To examine whether this action of DDX23 was specific to miR-21, DDX23-knockdown U87MG glioma cells were subjected to northern blot with a let-7-specific probe. Expression levels of the let-7 family are generally downregulated in numerous cancers (Lee et al., 2014), and low levels are correlated with poor prognosis on the contrary to miR-21 expression. As shown in Supplementary Fig. 4, DDX23 knockdown did not affect the levels of the precursor and mature let-7a miRNA together.
The effects of DDX23 depletion on the functions of mature miR-21 were tested by using a sensor that contained four consecutive miR-21 target sites (perfect match; Supplementary Fig. 5A) (Kim et al., 2011). As expected, downregulation of DDX23 derepressed the sensor activity mediated by mature miR-21 (Fig. 3C). Next, we examined the sensor activity of the mature miR-21 in 293T cells transfected with plasmids encoding the mature miR-21 sensor, GFP-DDX23 and pri-miR-21. As expected, DDX23 overexpression significantly downregulated the sensor activity mediated by the mature miR-21 (Fig. 3C).
The results presented in Fig. 3A–C strongly suggest that DDX23 is linked with the biogenesis of miR-21, especially at the primary-to-precursor miR-21 processing step (i.e. the cropping process by Drosha). To further examine whether DDX23 modulates pri-to-pre-miR-21 processing, we used our previously reported luciferase sensor to monitor whether DDX23 had any effect on microprocessor Drosha processing (Kim et al., 2011). We had previously showed that this sensor clearly responds to Drosha microprocessor activity, and that sensor activity was decreased in the presence of cellular modulators (proteins or RNAs) that modulate the Drosha processing step. In the present work, we found that DDX23 knockdown increased the activity of this luciferase-based sensor (Fig. 3D), whereas DDX23 overexpression had the opposite effect (Fig. 3D). This suggests that DDX23 potentiates pri-to-pre-miR-21 processing by regulating the activity of Drosha microprocessor.
Notably, we analysed miR-21 expression data from the REMBRANDT database and found correlations between the expression profiles of miR-21 and DDX23 in glioma patients. As shown in Fig. 3E, the miR-21 level in the DDX23-low group was lower than that in the DDX23-high group.
Collectively, these data show that DDX23 specifically affects miR-21 biogenesis by modulating the activity of Drosha.
DDX23 promotes invasion and proliferation of patient-derived glioma stem cells and cancer cell lines via modulating miR-21 biogenesis
As shown in Figs 2 and 3, we demonstrated potential role of DDX23/miR-21 signalling on modulation of invasion and proliferation of U87MG glioma cells in vitro and in vivo. Next, we hypothesized that this DDX23/miR-21 signalling is also conserved in patient-derived glioma stem cells (Singh et al., 2004; Lee et al., 2006; Hjelmeland et al., 2010; Jin et al., 2012; Yin et al., 2014). To test this possibility, we used small RNA northern blot analysis to examine the levels of the mature miR-21 in DDX23-knockdown glioma stem cells. Consistent with our hypothesis, DDX23-knockdown glioma stem cells showed reductions in the levels of mature miR-21 (Fig. 4A and D), furthermore, invasion assays revealed that DDX23-knockdown reduced the highly invasive phenotype of two glioma stem cells (Fig. 4B and E), suggesting that DDX23 is required for glioma stem cell invasion. Moreover, the proliferation of two glioma stem cells was suppressed by DDX23 knockdown (Fig. 4C and F).
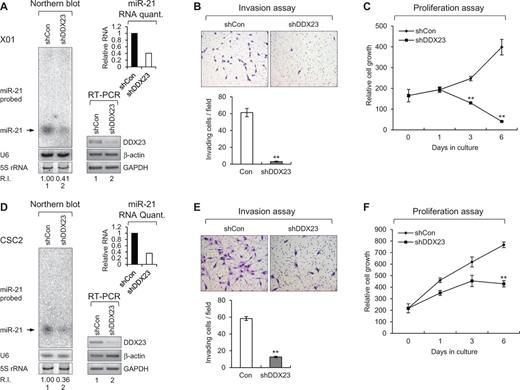
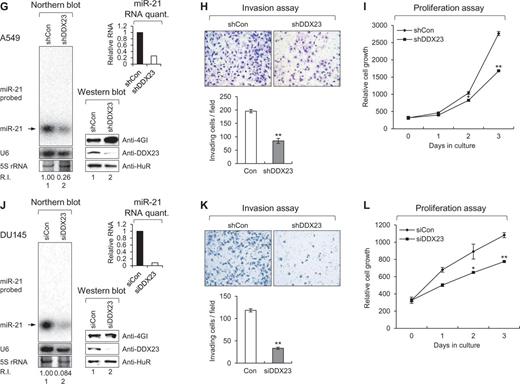
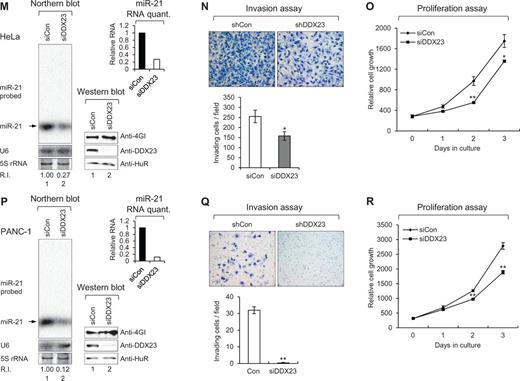
DDX23 promotes invasion and proliferation of patient-derived glioma stem cells (GSCs) and various cancer cell lines via modulating miR-21 biogenesis. (A, D, G, J, M and P) Specific knockdown of DDX23 induced downregulation of miR-21 in patient-derived glioma stem cells X01 (A), CSC2 (D), non-small cell lung cancer A549 (G), prostate cancer DU145 (J), cervical cancer HeLa (M) and pancreatic cancer PANC-1 (P) cell lines. Glioma stem cells X01, CSC2 and A549 were infected with control (shCon) or shDDX23 expressing lentiviral construct (shDDX23). Scrambled (siCon) or DDX23 specific (siDDX23) siRNA was transfected to the other cancer cell lines (DU145, HeLa and PANC-1). The level of endogenous miR-21 was monitored by northern blotting with miR-21-specific 32P 5′-end-labelled oligonucleotide probe. Arrow indicates position of mature miR-21. 5S rRNA was used as the loading control and U6 was used as hybridization control (lower left). The quantification of mature miR-21 is shown in graph (upper right). The experiments were repeated at least three times with similar results. The figures shown in A, D, G, J, M and P are representative. Specific knockdown of DDX23 was confirmed by semi-quantitative RT-PCR (X01 and CSC2) or western blot analysis (A549, DU145, HeLa and PANC-1) (lower right). (B, E, H, K, N and Q) Invasion assay was performed in patient-derived glioma stem cells and various cancer cell lines after specific knockdown of DDX23. Representative photos of invaded cells (upper), quantitative data of invaded cells (lower left), and all error bars in graph represent mean ± SEM (n = 3). *P < 0.05, **P < 0.01 (C, F, I, L, O and R) Proliferation assay was performed in patient-derived glioma stem cells and various cancer cell lines after specific knockdown of DDX23. All error bars in graph represent mean ± SEM (n = 3). *P < 0.05, **P < 0.01.
We also investigated potential conservation of this DDX23/miR-21 signalling other than glioma. Non-small cell lung (A549), prostate (DU145), cervical (HeLa) and pancreatic (PANC-1) cancer cell lines were tested for monitoring the possible role on invasion and proliferation after depletion of DDX23. As expected, all of these cells showed decreased miR-21, invasion, and proliferation by the specific knockdown of DDX23 (Fig. 4G–R). These data demonstrated that DDX23 is a critical regulator of invasion and proliferation in various cancers.
DDX23 promotes pri-miR-21 processing through an interaction with the Drosha microprocessor
As shown in Fig. 3, DDX23 promotes the cropping step of miR-21 biogenesis by regulating the microprocessor activity of Drosha. To examine whether DDX23 modulates Drosha activity through the formation of the DDX23-pri-miR-21-Drosha complex, we performed co-immunoprecipitation experiments for DDX23 and Drosha first. Extracts from 293T cells overexpressing FLAG-DDX23 were immunoprecipitated using an anti-FLAG M2 affinity gel and western blot with an anti-Drosha antibody. The results demonstrated that FLAG-DDX23 specifically bound to the endogenous Drosha microprocessor (Fig. 5A). This interaction was additionally confirmed by the co-transfection of 293T cells with plasmids encoding Myc-DDX23 and Drosha-FLAG, followed by co-immunoprecipitation experiments. As shown in Fig. 5B, Drosha-FLAG efficiently pulled down Myc-DDX23, but not β-actin or eIF4GI (an abundant mRNA-associated scaffold protein). These data suggest that DDX23 regulates pri-miR-21 processing via an interaction with the Drosha microprocessor.
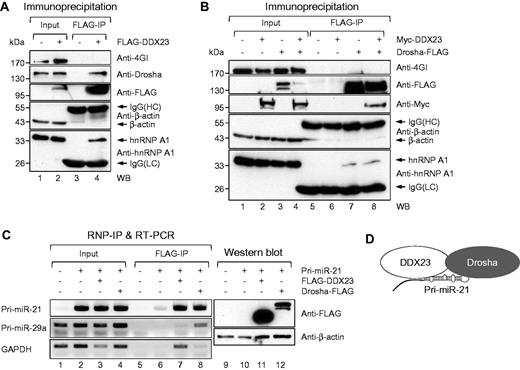
DDX23 specifically interacts with core microprocessor Drosha and pri-miR-21. (A) Plasmids encoding FLAG-DDX23 were ectopically expressed in 293T cells. FLAG-DDX23 was precipitated with anti-FLAG M2 affinity gel and then western blot performed with eIF4GI (negative binding control), Drosha, FLAG-tag, β-actin (negative binding control), and hnRNP A1 specific antibodies, respectively. IgG(HC) = IgG heavy chain; IgG(LC) = IgG light chain; IP = immunoprecipitation; WB = western blot. (B) Plasmids encoding Drosha-FLAG and Myc-DDX23 were ectopically expressed in 293T cells. Drosha-FLAG was precipitated with anti-FLAG M2 affinity gel and then western blot performed with eIF4GI (negative binding control), FLAG-tag, Myc-tag, β-actin (negative binding control) and hnRNP A1 specific antibodies, respectively. (C) Physical interaction between DDX23 and pri-miR-21. The plasmid encoding FLAG-DDX23 or Drosha-FLAG was ectopically expressed with pri-miR-21 RNA-encoding plasmid in 293T cells. RNA-protein complexes were precipitated with anti-FLAG M2 affinity gels. After immunoprecipitation of RNA-protein complexes, RNAs were isolated and used in RT-PCR reactions with specific oligomers for pri-miR-21, pri-miR-29a, and GAPDH. The PCR products were resolved on 1% agarose gel. Each protein expression was confirmed by western blot. (D) Proposed illustration shows that DDX23 cooperatively interacts with Drosha microprocessor complex and pri-miR-21, which facilitates pri-miR-21 processing.
To test whether DDX23 can directly bind to pri-miR-21, we used the anti-FLAG M2 affinity gel to perform ribonucleoprotein immunoprecipitation experiments against 293T cells expressing FLAG-DDX23 or control Drosha-FLAG, and then subjected the bound RNAs to semi-quantitative RT-PCR with specific primers against pri-miR-21, pri-miR-29 or GAPDH. With the exception of the Drosha-FLAG immunoprecipitation (positive control) experiment, we detected pri-miR-21 but not pri-miR-29a (Fig. 5C), indicating that DDX23 binds specifically to pri-miR-21 prior to the Drosha-mediated processing step. Together, these data strongly suggest that DDX23 specifically affects the processing of pri-miR-21 by cooperating with the Drosha microprocessor (Fig. 5D).
The helicase activity of DDX23 is essential for its effect on miR-21 biogenesis
The data in Figs 3 and 5 confirm that DDX23 facilitates pri-miR-21 processing via direct binding of pri-miR-21 and in conjunction with the Drosha microprocessor. As the activity of RNA helicases is important for spliceosome assembly and RNA processing, we hypothesized that this catalytic activity could also be essential for the DDX23-mediated processing of pri-miR-21. To test this possibility, we mutagenized the lysine 441 residue located in the highly conserved Walker A motif of human DDX23 to asparagine (K441N; Fig. 6A) (Teigelkamp et al., 1997). The mutation of this single amino acid did not alter the cellular localization pattern of DDX23 (data not shown). Overexpression of wild-type DDX23 [DDX23(WT)] repressed the luciferase activities of both primary and mature sensors around 75%, whereas the catalytically dead mutant (K441N) showed enhanced luciferase activities on the sensors (Fig. 6B), indicating that it had a dominant-negative effect on miR-21 biogenesis. Consistent with this finding, northern blot analysis showed that DDX23(WT) enhanced the generation of both precursor and mature miR-21, whereas DDX23(K441N) decreased these miR-21 levels below that seen in cells expressing control GFP (Fig. 6C). These results strongly suggest that the innate helicase activity of DDX23 is required for its ability to regulate miR-21 biogenesis. Consistent with this observation, expression of the DDX23(K441N) mutant suppressed the invasion (Fig. 6D) and proliferation (Fig. 6E) of U87MG glioma cells compared to cells expressing DDX23(WT).
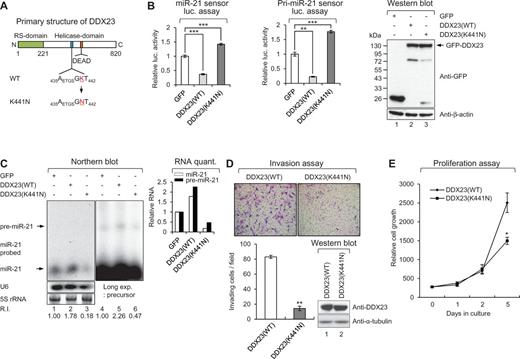
RNA helicase activity of DDX23 is essential for the miR-21 biogenesis, invasion and proliferation of U87MG glioma cells. (A) Schematic diagram of human DDX23 primary structure. Essential lysine residue in Walker A motif located in middle helicase domain was mutagenized as asparagine residue (K441N). RS-domain, arginine and serine rich domain; DEAD, aspartate-glutamate-alanine-aspartate. (B) DDX23(WT), but not K441N mutant affects the translation of mature and pri-miR-21 sensors. The mature or pri-miR-21 sensor was cotransfected into 293T cells along with the plasmid encoding GFP, GFP-DDX23(WT), or GFP-DDX23(K441N). The expression of GFP-DDX23s was confirmed by western blot. Data represent the mean values of at least three independent experiments performed in triplicate (***P < 0.001 and **P < 0.01). All error bars in the graph represent mean ± SEM and the P-value compares the control plasmid (GFP) to GFP-DDX23(WT), or GFP-DDX23(K441N). (C) K441N mutant has dominant negative effect on miR-21 biogenesis. Plasmids encoding GFP-DDX23(WT) or GFP-DDX23(K441N) were cotransfected with pri-miR-21 RNA encoding plasmid in 293T cells. After 48 h transfection northern blot was performed with miR-21-specific 32P 5′-end-labelled oligonucleotide probe. Arrows indicate positions of precursor (upper) and mature (lower) miR-21. 5S rRNA was used as the loading control and U6 was used as hybridization control. The quantification of mature and precursor miR-21 is shown in graph. R.I. = relative intensity. (D) Invasion assay was performed in U87MG glioma cells infected with DDX23(WT) or DDX23(K441N) expressing lentiviral construct. Representative photos of invaded cells (upper) and quantitative data of invasion cells (lower) were shown. All error bars in graph represent mean ± SEM. **P < 0.01. (E) Proliferation assay was performed in U87MG glioma cells infected with DDX23(WT) or DDX23(K441N) expressing lentiviral construct. All error bars in graph represent mean ± SEM. *P < 0.05.
The RNA helicase inhibitor ivermectin blocks DDX23-mediated miR-21 biogenesis and glioma cell proliferation
Based on our novel findings that DDX23 knockdown or abrogation of DDX23 RNA helicase activity can suppress the invasion and proliferation of glioma cells, we propose that DDX23 should be considered a novel therapeutic target for the treatment of glioma.
The action modes of the cellular DEAD-box RNA helicases are similar to those of the virally encoded helicases known to be essential for the translation and replication of the viral genome (Borowski et al., 2003; Mastrangelo et al., 2012; Mukherjee et al., 2012). To test the potential therapeutic targeting of DDX23, we assembled a number of chemical drugs that are known to inhibit viral helicases (Borowski et al., 2003; Mastrangelo et al., 2012; Mukherjee et al., 2012) and systematically tested their effects on our pri-miR-21 sensor system. Among the tested drugs, only the broad-spectrum anti-parasitic drug, ivermectin, inhibited the DDX23-mediated potentiation of pri-miR-21 processing (Fig. 7A and B). Ivermectin decreased the levels of both precursor and mature miR-21 in U87MG glioma cells, but did not affect DDX23 expression (Fig. 7C). Moreover, ivermectin completely suppressed cell proliferation of two patient-derived glioma stem cells (X01 and CSC2) and four cancer cell lines (A549, DU145, HeLa and PANC-1) including U87MG glioma cells (Fig. 7D–J).
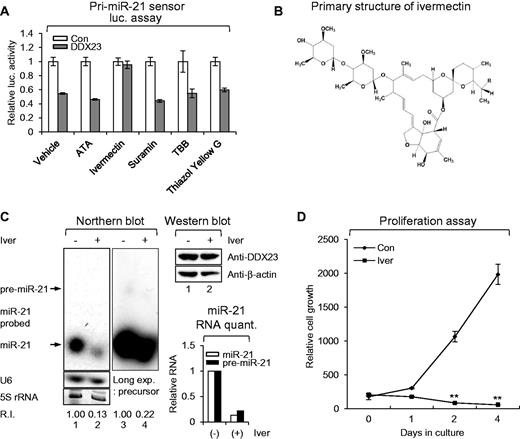
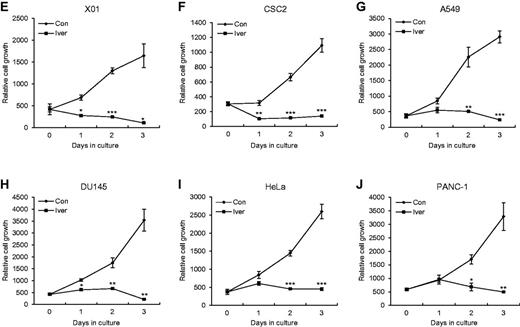
RNA helicase inhibitor ivermectin blocks proliferation of U87MG glioma cells via modulating DDX23/miR-21 signalling. (A) The pri-miR-21 sensor was cotransfected to 293 T cells along with GFP-DDX23 or control GFP encoding plasmid. Then, 100 µM of control DMSO (vehicle) or helicase inhibitors was treated on the cells for 24 h. Data represent the mean values of at least three independent experiments performed in triplicate. All error bars in graph represent mean ± SEM. (B) Primary structure of ivermectin. Component B1a, R = C2H5; Component B1b, R = CH3. (C) Ivermectin treatment induced downregulation of miR-21 in U87MG glioma cells. Ivermectin or control DMSO (10 μM) was treated on U87MG glioma cells for 24 h and then the level of endogenous miR-21 were monitored by northern blot. Northern blot was performed with miR-21-specific 32P 5′-end-labelled oligonucleotide probe. Arrows indicate positions of precursor (upper) and mature (lower) miR-21. 5S rRNA was used as the loading control and U6 was used as hybridization control (lower left). The quantification of precursor and mature miR-21 are shown in graph (lower right). Protein level of DDX23 and β-actin (loading control) were confirmed by western blot (upper right). The experiments were repeated at least three times with similar results. The figure shown in C is representative. R.I. = relative intensity. (D) Proliferation assay was performed in U87MG glioma cells treated or non-treated with ivermectin (25 µM). All error bars in the graph represent mean ± SEM. **P < 0.01. (E–J) RNA helicase inhibitor ivermectin abrogates proliferation of patient-derived glioma stem cells and various cancer cell lines. Proliferation assay was performed in glioma patient-derived glioma stem cells (X01, E; CSC2, F), non-small cell lung (A549, G), prostate (DU145, H), cervical (HeLa, I) and pancreatic (PANC-1, J) cancer cell lines treated with control DMSO (Con) or ivermectin (Iver, 25 µM). All error bars in graph represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To further demonstrate in vivo benefit of invermectin as a therapeutic agent, we performed in vivo treatment of ivermectin in mice. A previous report described that ivermectin shows neurotoxicity in mice at a dose > 0.5 mg/kg (Plumb, 2002); therefore, to provide enough amount of ivermectin in tumour area, we used intratumoural injection. As shown in Fig. 8A, the in vivo effect of ivermectin was substantial. When we used 3 mg/kg of ivermectin, we observed a ∼50% decrease of tumour size (Fig. 8A). Moreover, 10 mg/kg treatment showed near complete regression of tumours (Fig. 8A). Ki67 staining confirmed that glioma cell proliferation was decreased in ivermectin treated xenografts compared to control mice (Fig. 8B), suggesting that ivermectin suppressed glioma cell proliferation in vivo. Cleaved caspase 3 staining and TUNEL assay clearly showed ivermectin treatment induced apoptotic cell death of the xenograft in vivo (lanes ‘Cleaved caspase-3’ and ‘TUNEL’; Fig. 8B). Ivermectin also decreased the levels of both precursor and mature miR-21 in mouse xenograft but did not affect DDX23 expression (Fig. 8C). These data support ivermectin potentially induced regression of tumours in vivo via DDX23/miR-21 signalling and raised the possibility of ivermectin as potential therapeutic agent for human glioma.
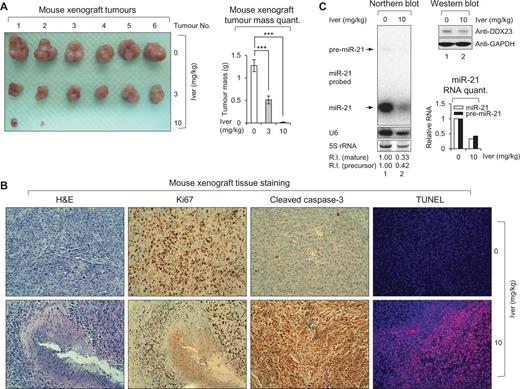
RNA helicase inhibitor ivermectin abrogates mouse xenograft growth in vivo. (A) Subcutaneous tumours isolated from ivermectin or its control vehicle-treated mice. Mice were subcutaneously injected with 5 × 106 U87MG glioma cells. These mice were administered with 3 mg/kg of ivermectin (3; middle line), 10 mg/kg of ivermectin (10; bottom line) or its control vehicle (0; upper line) as an intratumoural route. After 6 weeks, mice were sacrificed and then subcutaneous tumours were isolated. Total number of tumours for each group was six. Four tumours from 10 mg/kg of the ivermectin treated group were not detectable. Right: Graph represents mean tumour mass of each group. ***P < 0.001. (B) Histological analysis of subcutaneous tumour tissues isolated from 10 mg/kg of ivermectin (10; bottom panel) and its control vehicle (0; upper panel) treated mice. Isolated tissues were stained by haematoxylin and eosin (H&E), Ki67, cleaved caspase-3, and TUNEL, respectively. (C) Ivermectin treatment induced downregulation of miR-21 in vivo. Northern blot analysis of subcutaneous tumour tissues isolated from 10 mg/kg of ivermectin (10) and its control vehicle (0) treated mice. Northern blot was performed with miR-21-specific 32P 5′-end-labelled oligonucleotide probe. Arrows indicate positions of precursor (upper) and mature (lower) miR-21. 5S rRNA was used as the loading control and U6 was used as hybridization control (lower left). The quantification of precursor and mature miR-21 are shown in graph (lower right). Protein level of DDX23 and GAPDH (loading control) were confirmed by western blot (upper right). The experiments were repeated at least three times with similar results. The figure shown in C is representative. R.I. = relative intensity.
Discussion
The biogenesis of miR-21 was previously known to be deregulated in various cancers, but the relevant regulatory mechanism was largely unknown (Calin et al., 2005; Chan et al., 2005; Iorio et al., 2005; Diederichs and Haber, 2006; Roldo et al., 2006; Volinia et al., 2006; Lawrie et al., 2007; Meng et al., 2007; Wang et al., 2007). In the current study, we demonstrated that the upregulation of DDX23 in glioma is associated with miR-21 upregulation and poor patient prognosis. We further showed that ectopic overexpression of DDX23 promotes glioma cell invasion and proliferation, whereas DDX23 knockdown suppresses these parameters. In nude mice, DDX23-knockdown glioma cell xenografts yielded smaller tumour masses, less cell proliferation and longer survival than control glioma cell xenografts. Together, these results indicate that DDX23 plays an essential oncogenic role in glioma.
DDX23 is a RNA helicase whose cellular functions are generally determined by its target RNAs. Our group and others previously reported that miR-21 is elevated in gliomas, and associated with glioma cell invasion and proliferation (Gabriely et al., 2008; Papagiannakopoulos et al., 2008; Kwak et al., 2011). Thus, we herein hypothesized that miR-21 could be a target for DDX23-mediated regulation. Consistent with this, overexpression of DDX23 increased the levels of pre- and mature miR-21 while DDX23 knockdown decreased these parameters in a conventional cell line (U87MG) and patient-derived glioma stem cells (X01 and CSC2), suggesting that DDX23 may modulate the biogenesis of miR-21 in glioma. Moreover, miR-21 has been reported as an important oncogenic miRNA in various cancers such as breast, colon, lung, prostate, cervical and pancreatic cancers. As shown in Supplementary Fig. 1, breast invasive carcinoma and colon adenocarcinoma showed significant expression of DDX23 compared to the non-tumour tissue. As already reported these carcinomas showed significantly high level of miR-21 compared with non-tumour (Asangani et al., 2008; Yan et al., 2008; Moriyama et al., 2009; Folini et al., 2010; Yao and Lin, 2012; Yang et al., 2015). We further demonstrated DDX23/miR-21 signalling in these carcinoma-derived cell lines (A549, DU145, HeLa and PANC-1) and the signalling mechanism can serve as a potential pan-anti-cancer therapeutic target.
The biogenesis of miRNAs is a multi-step process (Kim et al., 2009), as follows: (i) miRNAs are transcribed in an RNA polymerase II-dependent manner; (ii) these primary miRNAs (pri-miRNAs) are capped and polyadenylated at their 5′ and 3′ ends, respectively; (iii) the pri-miRNAs are processed into precursor miRNAs (pre-miRNAs) by the Drosha microprocessor; and (iv) the pre-miRNAs are exported to the cytoplasm, where they are processed into mature miRNAs by the Dicer complex.
The gene encoding miR-21 is located within an intron of VMP1 (also known as TMEM49; Ribas et al., 2012), prompting us to speculate that the DDX23-mediated regulation of miR-21 biogenesis may reflect the splicing-related activity of DDX23. To exclude this possibility, we developed a sensor containing a ∼200 nucleotide portion of the miR-21-encoding sequence, including the pre-miR-21 sequence (Figs 3B and D and 6B and C). Using this sensor (which avoided the splicing effects of DDX23) to monitor pri-to-pre-miR-21 processing, we found that DDX23 regulates pri-to-pre-miR-21 processing by binding to the Drosha microprocessor.
We also questioned whether DDX23 might modulate Dicer processing during the biogenesis of miR-21. However, DDX23 is mainly localized in the nucleus (data not shown), making it unlikely to have an effect on cytoplasmic Dicer processing. Finally, the effect of DDX23 on the Drosha-mediated processing of miR-21 is consistent with other studies showing that members of the DEAD-box RNA helicase family, including DDX1 (Han et al., 2014), DDX5 (p68), and DDX17 (p72) (Fukuda et al., 2007), interact with the Drosha microprocessor complex to enhance the biogenesis of certain miRNAs.
Prior to this study, the DEAD-box RNA helicases were known to play crucial roles in miRNA processing and cancer, but it was largely unknown whether their helicase activity was directly required for such roles. Using mutational analysis, we herein show that the helicase activity of DDX23 is essential for glioma cell invasion, cell proliferation and miR-21 biogenesis. Thus, we propose that DDX23 could be a potential novel therapeutic target for the treatment of glioma.
Various RNA helicases prefer specific substrate RNAs and the role of a given RNA helicase can be classified as oncogenic or tumour suppressive depending on its substrate RNAs. Our present data clearly demonstrate that DDX23 acts as an oncogenic helicase by elevating the biogenesis of the oncogenic miRNA, miR-21. In the future, it may be possible to target miR-21 and other oncogenic substrates of DDX23, in the hopes of developing a novel therapeutic strategy against glioma.
Ivermectin is a broad-spectrum anti-parasitic drug that has been mainly used in humans to treat river blindness. A recent study found that ivermectin is an RNA helicase inhibitor with specific effects on the NS3 DEAD-box RNA helicase, which is indispensable for flavivirus replication (Mastrangelo et al., 2012). Furthermore, the avermectins, including ivermectin, reportedly inhibit the cell growth of various human cancers in vitro and in vivo (Drinyaev et al., 2004; Hashimoto et al., 2009; Sharmeen et al., 2010; Melotti et al., 2014). The potential activity of this agent against DDX23 and glioma had not been previously studied, but we herein show that ivermectin inhibits the pri-to-pre-miR-21 processing activity of DDX23 and decreases glioma cell proliferation. Furthermore, we also demonstrated that in vivo efficacy of ivermectin treatment for the glioma by using mouse xenograft model. Our results strongly suggest the possible application of ivermectin as an anti-cancer treatment.
In conclusion, the present study unequivocally demonstrates a new role for the DEAD-box RNA helicase, DDX23: it promotes miR-21 biogenesis in conjunction with the Drosha microprocessor (a key player in miRNA processing). Together with the existing reports, our findings provide insights into the role of DDX23 in tumorigenesis of various cancers. Moreover, in vivo treatment of ivermectin demonstrates pharmacological benefit of targeting DDX23/miR-21 signalling.
Acknowledgements
We especially thank V. Narry Kim (Seoul National University, Republic of Korea) for various expression constructs for the miRNA processing machinery. We are also grateful to Soeda Akio (Gifu University Graduate School of Medicine, Japan), Myung-Jin Park (Korea Institute of Radiological and Medical Science, Republic of Korea), Sang-Jin Lee (National Cancer Center, Republic of Korea), and Yun-Hee Kim (National Cancer Center, Republic of Korea) for GSCs X01, CSC2, prostate cancer cell line DU145 and pancreatic cancer cell line PANC-1, respectively.
Funding
This research was supported by grants from the National Cancer Center, Republic of Korea (NCC-1210041, NCC-1210042, NCC-1210043, NCC-1410080, NCC-1410290, NCC-1410300 and NCC-1510060), Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2011-0024692, NRF-2012R1A1A2009228, NRF-2013R1A1A2062555 and NRF-2014R1A1A2057118) and the Korean Health Technology R&D project, Ministry of Health & Welfare, Republic of Korea (A110878).
Supplementary material
Supplementary material is available at Brain online.
Abbreviation
References
Author notes
*These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}