




Abstract
Characterizing clinically relevant brain metastasis models and assessing the therapeutic efficacy in such models are fundamental for the development of novel therapies for metastatic brain cancers. In this study, we have developed an in vivo imageable breast-to-brain metastasis mouse model. Using real time in vivo imaging and subsequent composite fluorescence imaging, we show a widespread distribution of micro- and macro-metastasis in different stages of metastatic progression. We also show extravasation of tumour cells and the close association of tumour cells with blood vessels in the brain thus mimicking the multi-foci metastases observed in the clinics. Next, we explored the ability of engineered adult stem cells to track metastatic deposits in this model and show that engineered stem cells either implanted or injected via circulation efficiently home to metastatic tumour deposits in the brain. Based on the recent findings that metastatic tumour cells adopt unique mechanisms of evading apoptosis to successfully colonize in the brain, we reasoned that TNF receptor superfamily member 10A/10B apoptosis-inducing ligand (TRAIL) based pro-apoptotic therapies that induce death receptor signalling within the metastatic tumour cells might be a favourable therapeutic approach. We engineered stem cells to express a tumour selective, potent and secretable variant of a TRAIL, S-TRAIL, and show that these cells significantly suppressed metastatic tumour growth and prolonged the survival of mice bearing metastatic breast tumours. Furthermore, the incorporation of pro-drug converting enzyme, herpes simplex virus thymidine kinase, into therapeutic S-TRAIL secreting stem cells allowed their eradication post-tumour treatment. These studies are the first of their kind that provide insight into targeting brain metastasis with stem-cell mediated delivery of pro-apoptotic ligands and have important clinical implications.
Metastatic brain tumours typically occur as multiple foci, making them difficult to treat. Bagci-Onder, Du et al. report the development of an in vivo imageable breast-to-brain metastasis mouse model, as well as an efficient stem cell-based therapeutic strategy that allows targeted destruction of tumours and eradication of therapeutic stem cells post-treatment.
Introduction
Metastatic brain tumours are the most commonly observed intracranial tumours frequently occurring in patients with metastatic cancers, particularly from those of the lung, breast, and skin (melanoma) (Eichler et al., 2011). The incidence of brain metastasis has been estimated to be ∼35% of metastatic breast cancer patients (Lockman et al., 2010), which is often manifested with neurological symptoms and diagnosed with clinical imaging modalities (Steeg et al., 2011). Most patients have multiple metastatic lesions at the time of diagnosis, making surgery an inadequate therapeutic option on its own. Parallel therapeutic approaches include stereotactic radiosurgery, whole brain radiotherapy and chemotherapy (Kocher et al., 2011). However, the blood–brain barrier, which prevents the brain permeability of systemic therapies, and the negative side effects of radiotherapy pose challenges for the success of existing therapies and cause failure to improve overall patient survival (Lockman et al., 2010). Therefore, there is a considerable need for the development of novel tumour-specific therapies that aim to overcome the challenges of delivery and specificity issues.
Given the multistep and complex biological nature of brain metastasis, tumour models that recapitulate metastatic brain tumours are limited. There have been several studies that established tumour lines with metastatic potential (Bos et al., 2010) and specifically a clonal tumour population with brain-seeking properties have been described at a genomic level (Bos et al., 2009). A recent study reported a novel molecular mechanism for breast cancer cell infiltration into the brain by protection of cancer cells from death signals and allowing for efficient vascular co-option and colonization (Valiente et al., 2014). By expressing serpins, a family of protease inhibitors, metastatic tumour cells were able to adopt a unique defence mechanism and evade stroma-induced apoptosis. Therefore, activating the dormant, cell surface-mediated apoptotic mechanisms on tumour cells might be a favourable therapeutic approach for brain-metastatic tumours.
Tumor necrosis factor (ligand) superfamily, member 10 (TRAIL) belongs to the TNF receptor ligand family, which also includes TNF and FASLG (Wiley et al., 1995; Pan et al., 1997). It is well known that they are capable of inducing apoptosis in a number of cancer cells via the binding to their cognate receptors and the initiation of death receptor mediated signalling (French and Tschopp, 1999). Among them, TRAIL is a promising candidate for cancer therapies due to its capability of specifically targeting tumour cells while sparing normal cells (Stuckey and Shah, 2013). Therefore, it is of great interest to evaluate the strategy of targeting death receptor mediated apoptosis in metastatic tumours. However, in the case of brain neoplasms and metastases, local delivery of sufficient TRAIL ligand may be challenged by the presence of the blood–brain barrier.
We and others, have previously established a novel therapeutic approach for primary brain neoplasms. Using neural stem cells (NSC) and mesenchymal stem cells (MSC) engineered to express tumour-specific biomolecules, we have demonstrated the efficacy of stem cell-based anti-tumour therapies in both nodular and invasive glioblastoma models (Shah et al., 2008; Sasportas et al., 2009; Kauer et al., 2012; Stuckey and Shah, 2014). Among many other biological differences, one major difference between the primary and metastatic brain tumours is their localization and distribution in the parenchyma. Although primary brain tumours mostly arise at a single site and progress from there, metastases occur in several distinct spots giving rise to multiple metastatic foci of different sizes (Modha et al., 2005). With this being the major impediment for therapeutic success, using tumour-seeking stem-cell-based therapies might be effective in treating multiple metastatic tumour lesions in the brain. In this study, we first assessed in detail the nature of the metastatic breast cancer model, which was established through several rounds of brain selection of breast carcinoma cells (Bos et al., 2009). Using non-invasive bioluminescent and fluorescent imaging, we then assessed the temporal and spatial distribution of the metastases in the brain. We further explored the metastatropic properties of engineered NSCs and the therapeutic potential of TRAIL-secreting engineered neural stem cells (NSC-S-TRAIL) in this model. We establish that NSCs engineered to express metastatic tumour-specific biomolecules offer great potential in targeting brain metastasis.
Material and methods
Cell lines
MDA231-Br2Ma cells were kindly provided by Dr Joan Massague (Sloan Kettering Institute, NY) and cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% foetal bovine serum (FBS) and 1% penicillin-streptomycin as described (Bos et al., 2009). Primary mouse NSCs were obtained from Stem Cell Technologies and cultured in NeuroCult™ mouse neural stem cell basal medium (Stem Cell Technologies) supplemented with NeuroCult™ proliferation supplements, 2 μg/ml of heparin (Sigma), 20 ng/ml of human EGF (R&D Systems), and 20 ng/ml of human FGF2 (PeproTech) and penicillin/streptomycin as described (Bagci-Onder et al., 2011). Immortalized mouse NSCs, generated from embryonic cerebellum, were grown in DMEM supplemented with 10% FBS, 5% normal horse serum, sodium bicarbonate and sodium pyruvate as described (Tang et al., 2003). Human NSCs and human MSCs were used and grown as previously described (Kim et al., 2006; Shah et al., 2008; Sasportas et al., 2009).
Engineered viral vectors, viral packaging and transduction of cells
Lentiviral construct, Pico2-Fluc.mCherry, was gifted from Dr. Andrew Kung (Dana Farber Cancer Institute; Boston, MA) and packaged as described (Bagci-Onder et al., 2011). Pico2-Rluc.mCherry was constructed by replacing firefly luciferase (Fluc) with Renilla luciferase (Rluc). MDA231-BrM2a cells were transduced at a multiplicity of infection (MOI) of 2 in medium containing protamine sulphate (10 µg/ml). Retroviral vectors, MIGRI-TRAIL, MIGRI-TK-GFl (thymidine kinase-GFP Fluc) or MIGRI-GFl (GFP-Fluc) vectors are described elsewhere (Martinez-Quintanilla et al., 2013). NSCs and MSCs were transduced at multiplicity of infection of 5 using protamine sulphate. All cells were visualized by fluorescence microscopy for green fluorescent protein (GFP) or mCherry expression to confirm transduction. To prepare the conditioned medium, 200 000 mouse NSCs or NSC-S-TRAIL cells were seeded per well of a 6-well plate and cultured for 48 h before supernatant collection. ELISA was used to quantify secreted TRAIL in the conditioned medium from engineered NSCs as described previously (Bagci-Onder et al., 2013).
Cell viability assays
The effect of secreted TRAIL on cell viability was measured using CellTiter-Glo® (Promega) 24 h post-treatment as described (Bagci-Onder et al., 2013). All experiments were performed in triplicates. In co-culture experiments, the Fluc activity was used as an indicator of cell viability. Accordingly, MBr-FmC cells and mouse NSCs [NSC-GFP, NSC-S-TRAIL, NSC-TK (thymidine kinase), NSC-TR-TK (TRAIL-thymidine kinase)] were seeded at 1:1 ratio in 24-well (0.5 × 105/well; Costar). MBr-FmC cells were assessed for their viability using Fluc activity 48 h after co-culturing. In the other experiments, MBr-FmC were co-cultured with different ratios of engineered human NSCs or human MSCs and 48 h later Fluc activity was measured to indicate the viability of tumour cells.
Western blotting analysis
Following treatment with secreted TRAIL (24 h), MDA231-Br2Ma cells were lysed with NP40 buffer supplemented with protease (Roche) and phosphatase inhibitors (Sigma). For immunoblotting, 30 µg of harvested proteins from each lysate were resolved on 10% SDS-PAGE and immunoblotted with antibodies against caspase 8, caspase 9 (Cell Signaling), cleaved poly ADP-ribose polymerase (PARP) (Cell Signaling) or alpha tubulin (Sigma) and detected by chemiluminescence after incubation with horseradish peroxidase-conjugated secondary antibodies.
Creation of a brain metastasis mouse model
Athymic nude female mice (6–8 weeks of age, Charles River Laboratories) were anaesthetized with ketamine-xylazine and an incision was made to expose the right carotid artery. Using 8-0 suture, both common and internal carotid arteries were temporally ligated and a catheter connected to a 1 ml syringe was inserted into the external carotid artery to inject tumour cells. Two hundred thousand cells suspended in 100 µl phosphate-buffered saline (PBS) were slowly injected through the catheter. The external carotid artery was then permanently ligated under the dissecting scope (Olympus, SZX10) using fine surgical tools and blood circulation was restored by releasing both common and internal carotid blood flow. Mice were imaged for tumour cell presence a few days later and then periodically for tumour progression by bioluminescence. In a few experiments to assess tumour growth, tumour cells were injected directly into the brain parenchyma using stereotactic injection as described (Shah et al., 2008). The spatial distribution of brain metastases was examined by fluorescence microscopy on the brains of mice sacrificed at different time periods and as described below. All in vivo procedures were approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital.
Assessment of therapeutic potential of neural stem cells in tumour-bearing mice
To test both the metastatic tumour tracking (metastatropic) ability and efficacy of modified NSCs, NSCs (either expressing GFP, secreted TRAIL or Fluc) were injected into metastatic tumour-bearing mice by two different routes: (i) NSCs were stereotactically implanted into mice with established brain metastases into the brain parenchyma at a single site [200 000/5 µl PBS; from bregma, anteroposterior: −2 mm, mediolateral: 1.5 mm ventral (from dura): 2 mm] (Shah et al., 2008); or (ii) NSCs were injected via the left external carotid artery with microsurgical technique as described above. In both cases, 200 000 NSCs were injected and NSC distribution was assessed by immunofluorescence on brain sections obtained after injection, as described below. The efficacy of NSC application was assessed by measuring tumour growth using bioluminescence imaging. Mice were periodically sacrificed to examine histology, as described below.
Assessment of NSC-TR-TK in mouse brains
Two hundred thousand NSC-TR-TK-GFl cells were either intracranially or intracarotidly injected in nude mice. Seven days after intracranial injection, or 3 days after intracarotid injection, daily PBS or ganciclovir (75 mg/kg body weight) were administrated via intraperitoneal (i.p.) injection for 2 weeks. NSC fate was assessed by bioluminescence imaging at various time points. To assess the eradication of therapeutic NSC post-tumour treatment, MBr-RmC cells (50 000 cells/mouse) were intracranially injected in nude mice and tumour-bearing mice were injected intratumourally with NSC-TR-TK-GFl (50 000 cells/mouse) in the mouse brain. Daily PBS or ganciclovir were administrated via intraperitoneal injection after 4 days of NSC injection and the fate of NSCs was assessed by bioluminescence imaging at different time points.
Histology
Mice were sacrificed and brains were dissected and processed for immunohistochemistry as described (Shah et al., 2008). Ten-micrometre sections were assessed for mCherry and GFP expression representing tumour cells and NSCs, respectively. To assess the temporal and spatial distribution of tumour and stem cells, coronal sections were obtained along the anterior-posterior axis at 200 µm intervals. The composite images of each section were generated by stitching different individual images using Adobe Photoshop tools. The number of metastatic lesions was defined by measuring the number of mCherry-positive foci using ImageJ tools (NIH). Accordingly, each composite image was subjected to ImageJ particle analysis and the number of mCherry-positive particles per section was plotted. Caspase 3 staining was performed as described (Sasportas et al., 2009) using cleaved caspase-3 antibody (Cell Signaling). The number of caspase 3-positive puncta per tumour foci area was counted in multiple sections using ImageJ measurement tools. The average of these measurements per multiple sections was plotted. Endothelial cell staining was performed using anti-CD31 (Abcam). Whole-section images were acquired using a ×2 objective with a Olympus dissecting scope (SZX10) and processed using Adobe Photoshop. Higher magnification images were acquired with Olympus Digital Imaging Software (CellSens). Detailed section analysis was performed using confocal microscopy (LSM Pascal, Zeiss). Image quantification for caspase staining was performed using ImageJ and Adobe Photoshop measure tools.
Statistical analysis
Data were analysed by Student t-test when comparing two groups and ANOVA when comparing more than two groups. Data were plotted as mean ± SEM and differences were considered significant at P < 0.05. Survival curves were compared using the log-rank test. Analyses were done using GraphPad Prism 5.01.
Results
Characterization of brain metastasis in vivo
To establish an in vivo breast-to-brain metastatic model that can recapitulate the steps of metastatic progression and be imaged non-invasively, we engineered MDA231-BrM2a cells, which were previously isolated by several rounds of brain colonization from breast cancer (Bos et al., 2009), to express Fluc and mCherry using a bicistronic lentiviral vector (referred to as MBr-FmC; Supplementary Fig. 1A). Assessment of tumour growth after intracranial implantation of MBr-FmC confirmed their survival in the brain microenvironment (Supplementary Fig. 1B and Supplementary Data). To mimic a majority of the steps of metastatic colonization and blood vessel interactions, we injected MBr-FmC cells via the intracarotid artery in mice. Non-invasive bioluminescence imaging on tumour-bearing mice revealed exclusive brain metastases and the progression of tumour over 45 days, indicating tumour colonization throughout the brain (Fig. 1A and B). Assessment of mCherry fluorescence on coronal sections along the anterior-posterior axis 15, 25 and 45 days post-tumour cell injection, revealed distinct tumour foci that were detectable as early as 15 days post-tumour cell injection (Fig. 1C). The tumour cells distributed along all representative sections, and the number and size of metastases increased over time throughout the same coordinates correlating with increased bioluminescence. Quantification of the histology revealed a widespread distribution of micro- and macrometastases in the initial stages of metastatic progression (Day 15). The size of these metastatic lesions markedly increased at Day 25 and reached a maximally tolerated size at Day 45 post-MBr-FmC inoculation (Fig. 1D). Larger nodules developed haemorrhagic lesions, and astrogliosis was observed in the periphery of the tumours (Supplementary Fig. 1D–F). Furthermore, CD31 immunohistochemistry highlighting the brain vasculature revealed the full extravasation of tumour cells and the close association of tumour cells with blood vessels. Specifically, the tumour cells displayed distinct associations with vessels, such as vascular mimicry, neovascularization, subpial growth, vascular co-option (Fig. 1E), which constitute the hallmarks of brain metastasis. These results reveal that the intracarotid injection of breast to brain metastatic tumour cells result in the formation of a clinically-relevant mouse model that resembles the multi-foci metastases observed in the clinics.
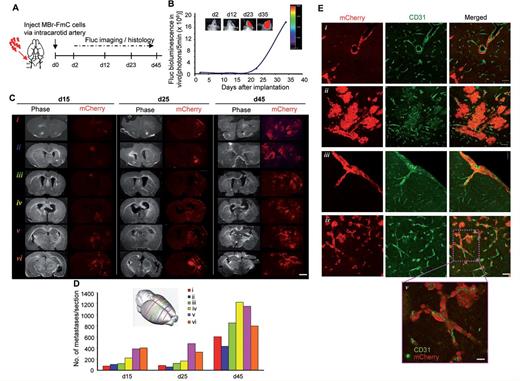
Imaging of brain metastases established with intracarotid artery injection reveals the tumour progression and blood vessel association in vivo. (A) Outline of the experiment. (B) Representative bioluminescent images of MBr-FmC tumours formed by intracarotid artery injection and plot showing the tumour growth of MBr-FmC over time. (C) Composite phase and fluorescent images (×2) of coronal brain sections generated from intracarotid injected tumours from different planes along the anterior-posterior axis (Scale bar = 2 mm, depicted with schematic in D, inset). (D) Quantification of metastatic tumour growth along different brain regions over time. (E) Representative fluorescent images from CD31 immunostaining on brain sections showing distinct hallmarks of brain metastasis. (i) vascular mimicry; (ii) neovascularization; (iii) subpial growth; (iv) vascular co-option (Scale bar = 100 µm); (v) close association of CD31 positive blood vessels and mCherry labelled tumour cells (Scale bar = 50 µm).
Stem cells migrate to metastatic tumour foci
We first tested two NSC lines for their ability to survive in vivo. A primary NSC line growing as neurospheres (referred to as primary NSCs) and an immortalized NSC line growing as monolayers were transduced to express GFl and implanted intracranially. Bioluminescence imaging showed that there was a direct correlation between NSC number and the Fluc bioluminescence imaging signal within the ranges tested (Supplementary Fig. 2A and Supplementary Data). In vivo survival studies revealed that immortalized NSCs survived markedly better than primary NSCs (Supplementary Fig. 2C). To test the migratory potential of NSC in metastatic brain tumours, we administered NSCs engineered to express GFP (NSC-GFP) intraparenchymally into metastatic brain tumour-bearing mice generated after intracarotid artery injection of MBr-FmC tumour cells (Fig. 2A). Intraparenchymal administration of NSCs resulted in detectable GFP-positive NSCs in the periphery of, or within the metastatic tumour foci throughout the brain (Fig. 2B and C). Histological examination of mCherry-labelled tumour cells and GFP-labelled NSCs in the brain sections revealed that NSCs were selectively located in the tumour-rich regions [Fig. 2B(i, iii, iv, v and vii)], but not in the tumour-free locations throughout the brain [Fig. 2B(ii and vi)]. Specifically, the migration of NSCs towards the metastatic deposits was observed for the parenchymal micro- and macrometastases [Fig. 2C(i)], leptomeningeal metastases [Fig. 2C(ii)], perivascular metastases [Fig. 2C(iii)], as well as cerebellar metastases [Fig. 2C(iv)], attesting to the highly tumoritropic migration ability of NSC towards metastatic brain tumours. Furthermore, CD31 immunohistochemistry analysis showed that migrating NSCs were present in or around tumour foci but not randomly distributed in the brain parenchyma or along blood vessels on their own (Supplementary Fig. 3). These results show that NSCs migrate toward metastatic foci in the brain, and this tumoritropic migration of NSC is unlikely guided by brain endothelium.
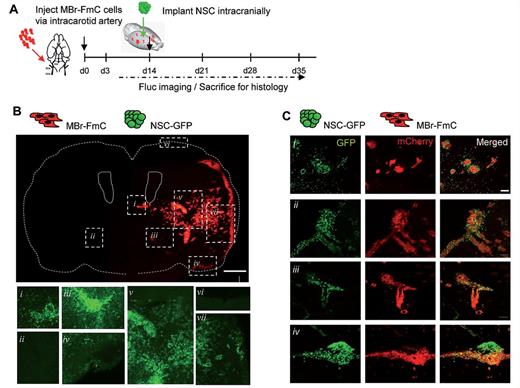
Interaction of NSC and metastatic tumour foci. (A) Outline of the experiment to study the association of engrafted NSCs with brain metastases after intracranial implantation of NSCs. (B) Representative composite fluorescent images of brain sections obtained at 2 weeks after NSC injection to an established brain metastasis model (Scale bar = 1 mm). Red, mCherry depicting tumour cells. Insets i–vii show the GFP labelled NSCs. (C) Representative fluorescent images showing NSC homing to established metastases. Green, NSC-GFP; Red, MBr-FmC tumour cells. (i) Small metastatic deposits surrounded by NSC; (ii) leptomeningeal metastasis (subpial) infiltrated by NSCs; (iii) perivascular metastasis infiltrated by NSCs; (iv) cerebellar metastasis accessed by NSCs (Scale bar = 50 µm).
NSCs secreting TRAIL home to metastatic foci and suppress brain metastasis growth in vivo
Based on the ability of NSCs to track metastastic deposits, we engineered NSCs to express a potent and secretable variant of tumour specific therapeutic agent, TRAIL (NSC-S-TRAIL). NSC-S-TRAIL secreted TRAIL (400 ng/ml) in the conditioned medium resulting in a significantly decreased MBr-FmC cell viability compared to the control NSC-GFP medium (Supplementary Fig. 4A). We also established co-cultures of MBr-FmC and NSC-S-TRAIL and showed a time-dependent decrease in MBr-FmC cell viability upon NSC-S-TRAIL treatment (Fig. 3A). Western blot analysis on the lysates of MBr-FmC treated with NSC-S-TRAIL revealed significant changes in initiator and effector caspases (caspases 8 and 3) and cleaved PARP as compared to the control treatment (Fig. 3B). To verify the in vivo potential of the therapeutic NSCs on metastatic tumour growth, we first admixed MBr-FmC cells and NSCs at 1:1 ratio and implanted mixed cells intraparenchymally. Bioluminescence imaging analysis showed a significant decrease in the MBr-FmC cell growth exposed to NSC-S-TRAIL, as opposed to the control NSC-GFP-exposed tumour cells that kept growing over time (Supplementary Fig. 4B). Next, we tested the therapeutic potential of stereotactically implanted NSC-S-TRAIL in our brain metastasis model. A marked suppression of metastatic tumour growth was observed upon NSC-S-TRAIL treatment as compared to NSC-GFP treatment (Fig. 3C). The whole brain sections collected at Day 40 revealed the growth and distribution of mCherry labelled tumour cells surrounded and intercalated by GFP-labelled control NSC. In contrast, the NSC-S-TRAIL treated tumours were less abundant confirming the homing and therapeutic efficacy of NSC-S-TRAIL in the metastasis model (Fig. 3D and E). A significantly higher cleaved caspase 3 expression on the brain sections confirmed NSC-S-TRAIL-mediated tumour cell apoptosis (Fig. 3F). These results show that NSC-secreted secreted TRAIL induce caspase 3-mediated apoptosis in metastatic tumour cells both in vitro and in vivo.
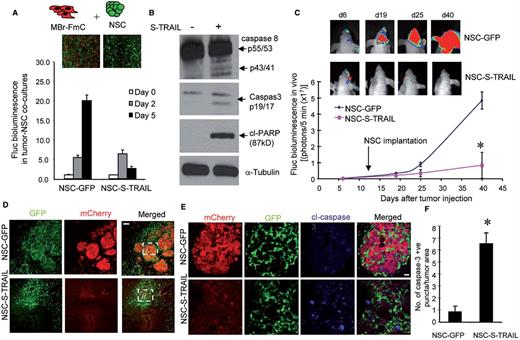
NSCs secreting TRAIL home to metastatic foci and eradicate brain metastases in vivo. (A) Viability of MBr-FmC cells in tumour-NSC co-cultures as measured by Fluc activity in vitro. The effects of NSCs engineered with GFP or secreting TRAIL are shown. (B) Western blot analysis of caspase 8, caspase 3, cleaved PARP1 and tubulin in MBr-FmC lysates collected upon treatment with conditioned medium from NSC-GFP (-, without secreted TRAIL in the medium) or NSC-S-TRAIL ( + , with secreted TRAIL in the medium). (C) Top: Representative bioluminescent images of metastatic MBr-FmC tumours established with intracarotid injection and administered with NSC-GFP or NSC-S-TRAIL on Days 6, 19, 25 and 40 after tumour cell injection. Bottom: Plot showing Fluc activity of MBr-FmC tumours treated with NSC-GFP (n = 3 mice) or NSC-S-TRAIL (n = 5 mice). Arrow points to the time of intracranial NSC injection (14 days after tumour cells injection). (D) Representative fluorescent images of coronal brain sections obtained from tumour bearing mice treated with NSC-GFP or NSC-S-TRAIL. Red, MBr-FmC cells; Green, NSC-GFP or NSC-S-TRAIL (Scale bar = 100 µm). (E) Representative confocal images of cleaved caspase 3 immunohistochemistry of metastatic tumours surrounded by NSC-GFP (top) or NSC-S-TRAIL (bottom) obtained at Day 40. Red, MBr-FmC cells; Green, NSC-GFP or NSC-S-TRAIL; Blue, cleaved caspase 3 (Scale bar = 25 µm). (F) The number of activated caspase 3 positive cells in E was calculated. (n = 5 regions/group counted for blue puncta and red area).
Next, we tested the survival and distribution of NSCs in a clinical relevant strategy wherein we administered NSCs or NSC-S-TRAIL via circulation (by intracarotid artery injection on the contralateral site of tumour inoculation) in the breast-to-brain metastasis model. To image tumour cells and NSC simultaneously, we created lines MBr-RmC (MDA231-BrM2a expressing Rluc and mCherry) and NSC-GFl (NSC expressing both Fluc and GFP). Dual Rluc and Fluc bioluminescence imaging confirmed the presence of both metastatic tumours and NSCs in the brain (Fig. 4A and B). Furthermore, representative fluorescent images revealed that intracarotid injection of NSCs resulted in detectable GFP-positive NSCs in the periphery of, or within the metastatic tumour foci in the brain (Fig. 4C). Next, we tested the therapeutic efficacy of NSC-S-TRAIL administered via circulation in the brain metastasis model (Fig. 4D). Bioluminescence imaging showed a significant suppression of metastatic tumour growth in the brain of NSC-S-TRAIL treated group compared with NSC-treated group (Fig. 4E). A significant survival benefit of the metastatic tumour-bearing mice was observed in the NSC-S-TRAIL-treated group compared to the NSC group or control group (Fig. 4F). Taken together, our results show that NSCs engineered to secrete TRAIL demonstrate efficacy in breast-to-brain metastasis by homing to metastatic deposits and inducing tumour-specific apoptosis in the brain.
![Intravascular administration of NSC-S-TRAIL in metastatic tumour bearing mice have anti-tumour effects. (A) Outline of the experiments to investigate the survival and distribution of NSC in the brain of metastatic tumour bearing mice. (B) Representative dual bioluminescent images of both MBr-RmC and NSC-GFl cells administered via intracarotid injection. Rluc imaging depicting tumour cells [Day 0 (d0) representing 14 days after tumour cell injection] and Fluc imaging depicting NSC (2, 5 and 14 days after NSC injection). Plot showing the Fluc bioluminescence of NSC-GFl cells over time (n = 3 mice). (C) Representative fluorescent images showing NSC surrounded or located within metastatic tumour foci 7 days after intracarotid administration (Scale bar = 50 µm). (D) Outline of experiments to study the efficacy of NSC-S-TRAIL administered intracarotidly in metastatic tumour-bearing mice. (E) Plot showing the Fluc bioluminescence of MBr-FmC tumours without treatment (control, n = 5 mice) or treated with NSC (n = 11 mice) or NSC-S-TRAIL (n = 12 mice). Inset: representative bioluminescent images of metastatic MBr-FmC tumours established with intracarotid injection and treated with NSC or NSC-S-TRAIL on Day 35. (F) Kaplan-Meier survival curves of metastatic breast tumour bearing mice without treatment as control (n = 7 mice) or treated with NSC (n = 6 mice) or NSC-S-TRAIL (n = 7 mice, P = 0.021 in NSC and NSC-S-TRAIL comparison, log-rank test).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/138/6/10.1093_brain_awv094/2/m_awv094f4p.jpeg?Expires=1716385230&Signature=T2phayobYY~c-ALqIAyvFdu8EP1FDJvAqy6Ad4~6mhrGT71ikacYd2phtCyyZmu3ZKZnloMi8RcB1nPlKmacx1rFZA7X150j7IcCx6eu7iLEshitNwRK71GffZv9um2c3qdmytmjb9Gehjph8afVq1mkh0ua5DvPDvns28s81JfVQSu8WHK1aJSMYehGNRn24cfQCF6d-WRnBejkclRYllBcB98TjqdJPJ~x3OJE7225ToHifboReN-ZpnKjRY6RwdozJ6~axt~a54AvQJKQeWGOTlv3bHr2bugYP5s5fAHt4fecwP-Th6NR0GF-RMqHler8qEDzc8F1KEpMZZXgHw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Intravascular administration of NSC-S-TRAIL in metastatic tumour bearing mice have anti-tumour effects. (A) Outline of the experiments to investigate the survival and distribution of NSC in the brain of metastatic tumour bearing mice. (B) Representative dual bioluminescent images of both MBr-RmC and NSC-GFl cells administered via intracarotid injection. Rluc imaging depicting tumour cells [Day 0 (d0) representing 14 days after tumour cell injection] and Fluc imaging depicting NSC (2, 5 and 14 days after NSC injection). Plot showing the Fluc bioluminescence of NSC-GFl cells over time (n = 3 mice). (C) Representative fluorescent images showing NSC surrounded or located within metastatic tumour foci 7 days after intracarotid administration (Scale bar = 50 µm). (D) Outline of experiments to study the efficacy of NSC-S-TRAIL administered intracarotidly in metastatic tumour-bearing mice. (E) Plot showing the Fluc bioluminescence of MBr-FmC tumours without treatment (control, n = 5 mice) or treated with NSC (n = 11 mice) or NSC-S-TRAIL (n = 12 mice). Inset: representative bioluminescent images of metastatic MBr-FmC tumours established with intracarotid injection and treated with NSC or NSC-S-TRAIL on Day 35. (F) Kaplan-Meier survival curves of metastatic breast tumour bearing mice without treatment as control (n = 7 mice) or treated with NSC (n = 6 mice) or NSC-S-TRAIL (n = 7 mice, P = 0.021 in NSC and NSC-S-TRAIL comparison, log-rank test).
Elimination of therapeutic stem cells by HSV-TK
To translate NSC-S-TRAIL therapy for brain metastasis into clinics, it is critical to follow the long-term fate of NSCs post-tumour treatment and ultimately, selectively eradicate therapeutic NSCs to avoid continuous access of therapeutic agent to normal brain tissue and to circumvent any malignant transformation of NSCs. To address this, we engineered NSC-S-TRAIL to further express herpes simplex virus thymidine kinase (HSV-TK) and GFP-Fluc (GFl) (NSC-TR-TK-GFl) and tested the efficacy of thymidine kinase suicide therapy in vitro. As revealed by both Fluc bioluminescence imaging and GFP immunohistochemistry, administration of ganciclovir eliminated modified NSC in vitro (Supplementary Fig. 5A). We next confirmed that NSCs retain their capacity to reduce tumour cell viability, without themselves being killed (Supplementary Fig. 5B). Further we tested the efficacy of ganciclovir to eradicate NSCs in non-tumour bearing brain. As revealed by Fluc bioluminescence imaging, NSCs were substantially eradicated by thymidine kinase suicide therapy, either implanted via the intracranial (Fig. 5A) or intracarotid route (Fig. 5B). Next, we tested the eradication of therapeutic NSCs post-tumour treatment in vivo. NSC-TR-TK-GFl were intracranially injected into MBr-RmC tumour-bearing brains. Fluc bioluminescence imaging revealed the eradication of therapeutic NSC-TR-TK-GFl after ganciclovir administration compared to control group treated with PBS (Fig. 5C). Fluorescent images of brain sections collected from tumour-bearing mice (before/after ganciclovir treatment) confirmed the elimination of MBr-RmC tumour cells by NSC-TR-TK-GFl administration (Supplementary Fig. 5C). These results reveal that therapeutic stem cells can be eradicated both in vitro and in vivo post-tumour treatment.
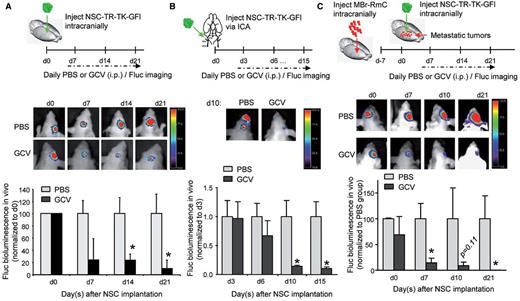
Therapeutic NSC can be eradicated post-tumour treatment in vivo. (A) Top: Outline of the experiment to test the eradication of intracranially injected NSC by ganciclovir (GCV) administration. Representative bioluminescent images of mice intracranially administered with NSC-TR-TK-GFl cells with or without ganciclovir treatment. Plot showing the Fluc activity of NSC-TR-TK-GFl cells in vivo (n = 3 mice per group). (B) Top: Outline of the experiment to test the eradication of intracarotidly injected NSCs by ganciclovir administration. Representative bioluminescent images of mice intracarotidly administered with NSC-TR-TK-GFl cells and treated with PBS or ganciclovir. Plot showing the Fluc activity of NSC-TR-TK-GFl cells in indicated groups in vivo (n = 5 mice per group). (C, top) Outline of the experiment to test the eradication of NSC post-tumour treatment. Representative bioluminescent images of mice intracranially administered with NSC-TR-TK-GFl with or without ganciclovir treatment. Plot showing the Fluc activity of NSC-TR-TK-GFl cells in vivo (n = 4 mice per group).
Human NSCs and MSCs expressing secreted TRAIL and HSV-TK have anti-tumour effects
To make our therapeutic strategy more clinically relevant, we used human NSCs and human MSCs and tested the efficacy of the TRAIL/thymidine kinase system on metastatic tumour cells. We engineered human NSCs and MSCs to express secreted TRAIL (hNSC-TR/hMSC-TR) or co-express secreted TRAIL and HSV-TK (hNSC-TR-TK/hMSC-TR-TK), and established co-cultures of MBr-FmC with different proportions of modified human NSCs and MSCs. Fluc bioluminescence imaging data showed a dose-dependent decrease of MBr-FmC cell viability within the different proportions of therapeutic human NSCs and MSCs tested (Fig. 6A and C). Furthermore, the administration of ganciclovir successfully eradicated hNSC-TR-TK/hMSC-TR-TK in vitro (Fig. 6B and D). These results reveal that human MSCs and NSCs, like their mouse counterparts, have the same efficacy of killing breast-to-brain metastatic tumour cells and can be eliminated post-ganciclovir treatment.
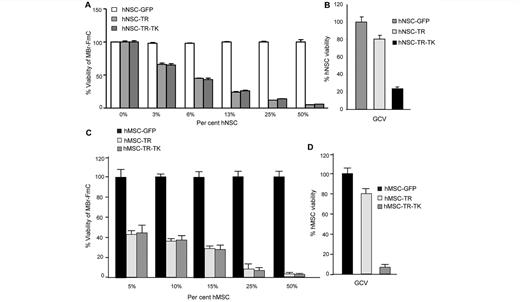
Human NSC and human MSC secreting TRAIL/HSV-TK kill metastatic tumour cells in vitro. (A) Plot showing the viability of MBr-FmC cells co-cultured with engineered human NSCs expressing TRAIL (TR), both TRAIL and HSV-TK (TR-TK) or GFP as measured by Fluc activity in vitro. (B) Plot showing the viability of modified human NSCs treated with ganciclovir (10 µg/ml). (C) Plot showing the viability of MBr-FmC cells co-cultured with engineered human MSC expressing TRAIL (TR), both TRAIL and HSV-TK (TR-TK) or GFP. (D) Plot showing the viability of modified human MSCs treated with ganciclovir (10 µg/ml).
Discussion
In this study, we first developed and extensively characterized an imageable model of clinically-relevant breast-to-brain metastasis that displays various features of brain metastasis. We then show the ability of engineered NSCs to home to metastatic brain tumours in this model. Furthermore, we show that therapeutic NSC-S-TRAIL significantly suppresses metastatic tumour growth and prolongs the survival of mice bearing metastatic breast tumours in the brain. We also demonstrate that the incorporation of pro-drug converting enzyme, HSV-TK into therapeutic NSC-S-TRAIL allows the eradication of NSCs post-tumour treatment.
Based on case series from the 1960s and 1970s, the incidence of clinically evident brain metastasis among women with stage IV breast cancer is estimated to be 10–16%. These figures underestimate the true incidence, given that brain metastases are found in 30% of patients at autopsy (Lin et al., 2004). Although a number of previous studies have made significant advances in our understanding of the biology of brain metastasis, it is still of utmost importance to use the most suitable model that mimics the clinical settings. Most of these studies have used intracranial injection of tumour cells directly to the brain parenchyma (Eichler et al., 2011), which does not mimic the actual clinical settings. There are multiple steps to metastatic colonization of the brain, namely, initial arrest of tumour cells at the brain capillaries, extravasation, continuation of perivascular position, vessel co-option, micrometastatic growth, and macrometastatic growth. To mimic a majority of the steps of metastatic colonization and blood vessel interactions, we created an imageable model of breast-to-brain metastasis by injecting tumours cells via the intracarotid artery in mice. Our histological examination of tumour cell–endothelial cell interactions on the mice brains inoculated with MBr cells, displayed all these features in consistency with a report that assessed the single steps of brain metastasis with another cell line model (Kienast et al., 2010). As the wide localization of multiple lesions is a major impediment in the treatment of such metastasis, it is crucial to use models that display this feature to develop effective therapies.
One of the major causes of therapeutic failure in metastatic brain cancer is the insufficient delivery of drugs across the blood–brain barrier (Eichler et al., 2011). As most metastatic lesions in the brain bear non-leaky vasculature, the success of systemic therapies is only marginal. Therefore, on-site delivery of tumour-specific therapeutics to the metastatic lesions would offer great potential for these tumours. We and others have previously documented a novel approach of delivering cytotoxic therapies to primary brain tumours, specifically glioblastomas, by using stem cells as cellular vehicles (Park et al., 2002; Tang et al., 2003; Shah et al., 2004, 2005; Schmidt et al., 2005; Corsten and Shah, 2008; Zhao et al., 2008; Kauer et al., 2012; Stuckey and Shah, 2014). NSCs that are engineered to express tumour-specific cytotoxic biomolecules, such as TRAIL; or prodrug-converting enzymes, such as carboxyl esterase, have been shown to home to and eradicate glioblastomas in preclinical mouse models (Yip and Shah, 2008; Stuckey and Shah, 2014). Indeed, this novel approach is now on its way to phase I and II clinical trials for recurrent glioblastomas.
The cell-of-origin of metastatic brain tumours resides in a distant organ, as opposed to being in the brain, as for primary brain tumours; thus, the biology of metastatic tumours is markedly different than that of primary brain tumours. Although armed NSCs are now considered to be efficacious in primary brain tumours, their potential in metastasis models is vaguely characterized. There have been a few reports on the use of stem cells for targeting metastasis in the brain (Seol et al., 2011; Kaneko et al., 2015). These studies either used the direct implantation of stem cells in the brain and did not thoroughly assess the interaction of engrafted stem cells with metastatic lesions by utilizing different imaging tools. To our knowledge, ours is the first study of its kind to demonstrate a novel stem cell-based approach for established brain metastasis. We now show that intracarotid injection of tumour cells display all the features of metastasis and that NSCs, either engrafted directly to brain or administered via circulation, have immense ability to track metastatic tumour lesions. Therefore, ‘arming’ NSCs with anti-tumour biomolecules offers great potential to eradicate these multifocal lesions.
Previous work showed that the infiltrating metastatic breast cancer cells that survived within the brain were shielded from death receptor-mediated apoptosis signalling (Valiente et al., 2014), suggesting that death receptor signalling is a promising target for breast-to-brain metastasis. We also found that the expression level of TNFRSF10B (previously known as DR5) was extremely high in the metastatic MBr cells (unpublished data) and TRAIL specifically killed the tumour cells instead of the normal cells. We thus use TRAIL as an anti-tumour biomolecule to induce metastatic cell apoptosis via death receptor mediated signalling. Further, we demonstrated that NSC-delivered secreted TRAIL induces apoptosis in the brain-metastatic breast cancer cell line MBr in vitro and in vivo. Additionally, we have shown that direct implantation of NSCs at a single region in the brain parenchyma was sufficient in eradicating many metastatic lesions spread throughout the brain in our model. When administered through the blood flow, NSC-S-TRAIL extravasated at tumour-rich sites and eradicated tumour lesions. This also conferred a significant survival advantage to mice, further revealing the in vivo efficacy of NSC-delivered secreted TRAIL for metastasis in the brain. This approach could be further improved by engineering NSCs to be more permeable through the brain endothelium by introducing the markers identified for transendothelial migration, such as, ST6GALNAC5 (Bos et al., 2009). All in all, regardless of the route of administration, NSC-S-TRAIL displayed significant efficacy in metastatic lesions.
As also studied for other cancer types, TRAIL or other death receptor agonists have received considerable attention because of their tumour specificity (Wiezorek et al., 2010). It will be of huge interest to assess the response of other metastatic cell lines to NSC-delivered secreted TRAIL both in vitro and in vivo. Whether TRAIL sensitivity is inherently present in the cancer origin or is acquired during metastasis to the brain might dictate the tailoring of metastatic tumour-specific therapies. In the case where the metastatic lesions are not NSC-S-TRAIL-responsive, it might be possible to use combinatorial therapies as we have shown for primary brain cancers (Bagci-Onder et al., 2011, 2013). It will be also of interest to assess the efficacy of standard breast cancer chemotherapeutics in combination with NSC-delivered secreted TRAIL in metastatic brain tumours.
To avoid possible de novo tumour formation and ensure safety of the use of stem cells, it would be best to remove residual stem cells after they have carried the therapeutics to the tumours. To eliminate therapeutic NSCs from the brain, we engineered NSCs with a suicide gene system where the administration of pro-drug ganciclovir was sufficient to remove the HSV-TK expressing NSCs from the brain after treatment. Tumour elimination and eradication of therapeutic cells are expected to result in a burden of dead cells in the tumour microenvironment. This might possibly activate gliosis and microglial activation contributing to the clearance of cellular debris, as reported previously for brain injury models (Kim et al., 2015). Although the long-term consequences of NSC-TK based therapies remain to be explored in detail, our study offers a novel future approach, where NSCs engineered to simultaneously express blood–brain barrier permeability genes, pro-apoptotic molecules, and suicide genes might reveal efficacy against brain metastases.
Funding
This work was supported RO1 CA138922 (K.S.), James McDonnell Foundation (K.S.).
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- Fluc
firefly luciferase
- MSC
mesenchymal stem cells
- NSC-S-TRAIL
neural stem cells engineered to secrete TRAIL
References
Author notes
*These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}